Palabras clave
INTRODUCCIÓN
La hipertensión pulmonar complica la evoluciónde muchos niños y adultos con cardiopatías congénitas (CPC). El aumento de la presión pulmonarasociado a la CPC es secundario a un aumento delflujo sanguíneo pulmonar o a la elevación de laspresiones poscapilares. La hipertensión arterial pulmonar (HAP) se asocia en la inmensa mayoría delos casos a cortocircuitos congénitos.
A pesar de los importantes avances que se hanrealizado en el conocimiento de la regulación del lecho vascular pulmonar y las lesiones endotelialespulmonares que dan lugar a la enfermedad vascularpulmonar, y a pesar de los avances en las reparaciones quirúrgicas y del descubrimiento de posiblestratamientos para los periodos preoperatorio ypostoperatorio, la hipertensión pulmonar continúacomportando una mortalidad y una morbilidad significativas en los pacientes con CPC.
Uno de los aspectos más importantes que es preciso definir es la formulación exacta utilizada paradefinir la enfermedad en el contexto de la HAP asociada a las CPC. Si se usa la definición hemodinámica de la HAP (presión arterial pulmonar [PAP]media > 25 mmHg)1, casi todos los pacientes tienenhipertensión pulmonar en presencia de un cortocircuito izquierda-derecha amplio y sin restricción,pero lo que tiene importancia en esta situación es elgrado de las lesiones vasculares pulmonares y loque podría denominarse la enfermedad vascularpulmonar (EVP). De hecho, un paciente con unflujo sanguíneo pulmonar elevado y una resistenciavascular pulmonar (RVP) baja cumplirá los requisitos exigidos para un diagnóstico de HAP, pero sepuede tratarlo de forma definitiva con el cierre quirúrgico de la comunicación. Por el contrario, un paciente con un flujo sanguíneo pulmonar bajo, cianosis con inversión del cortocircuito (derecha aizquierda) y RVP alta y el denominado síndrome deEisenmenger (SE) no obtendrá un efecto beneficioso con el cierre quirúrgico, que incluso está contraindicado, pero sí podrá obtenerlo con el uso delos nuevos tratamientos específicos para la HAP.
La reciente introducción de las terapias dirigidasa otras formas de HAP ha motivado un renovadointerés por la hipertensión pulmonar asociada aCPC, sobre todo en lo relativo a la forma másavanzada de esta, el SE2.
La situación concreta de la fisiología de un soloventrículo es también de gran interés, puesto que unaumento siquiera mínimo de la RVP puede impedirla operación de Fontan o llevar al fallo de esta circulación3.
En esta revisión se resume el conocimiento actualsobre la hipertensión pulmonar asociada a las CPCcaracterizadas por cortocircuitos congénitos. Se comentará de manera específica la HAP preoperatoriay postoperatoria, el tratamiento del SE y la situación concreta de la circulación de Fontan.
EPIDEMIOLOGÍA Y CLASIFICACIÓN
Hay una gran variedad de CPC que pueden conducir a la HAP, pero el grupo más importante es elformado por las lesiones con un cortocircuito izquierda-derecha. Dicho grupo incluye muchos defectos congénitos diferentes que presentan evoluciones diferentes, y ello tiene importancia.
Los avances realizados en cardiología pediátricay en cirugía han hecho aumentar el número de pacientes con CPC que sobreviven hasta la edadadulta, y han ayudado a prevenir la aparición delSE en muchos pacientes de los países occidentales,con lo que se ha producido una reducción deaproximadamente un 50% en la prevalencia a lolargo de los últimos 50 años. Alrededor del 5% delos adultos con CPC acaban sufriendo HAP4. Laprevalencia de la HAP en las CPC se ha estimadoentre 1,6 y 12,5 millones de adultos, de los que un25-50% presenta SE5. Sin embargo, sigue creciendoel número de pacientes con malformaciones caracterizadas por la denominada fisiología de ventrículoúnico, que requieren un abordaje quirúrgico especial (anastomosis cavopulmonar parcial o total).Aunque no presenten hipertensión pulmonar atenor de la definición clásica, estos pacientes puedentener lesiones vasculares pulmonares que impidanla intervención quirúrgica o hagan que esta comporte un riesgo elevado de morbilidad y mortalidad.
Los cambios estructurales de la circulación pulmonar en todas las formas de HAP, incluido el SE,son cualitativamente similares, aunque existen algunas diferencias en la distribución y la prevalenciade las alteraciones anatomopatológicas con las diferentes etiologías subyacentes. Según la clasificación,la hipertensión pulmonar debida a CPC se agrupacon la HAP idiopática/heredable, la HAP asociadaa fármacos, la HAP asociada a enfermedades deltejido conjuntivo y la HAP relacionada con elVIH6. Sin embargo, como se ha mencionado antes,la CPC es un grupo complejo de trastornos quepueden diferir de otras formas de HAP en lo relativo a la anatomía cardiaca, la hemodinámica y lahistoria natural de la enfermedad7,8. Esta es una delas razones por las que los expertos han intentadodesarrollar progresivamente una subclasificaciónque permita definir mejor a los pacientes con HAPCPC8-10. En las subclasificaciones se ha tenido encuenta varios factores importantes para describirmejor las lesiones, pero también factores que sonimportantes en el desarrollo de la EVP, como el tipo y el tamaño de los defectos congénitos, la hemodinámica, la presencia de anomalías cardiacasadicionales y el estado de la reparación (no reparado, paliado o reparado). Partiendo de estas sugerencias y con un mejor conocimiento de la enfermedad, en el último congreso mundial dehipertensión pulmonar se han introducido variasmodificaciones que permiten realizar una clasificación anatomopatológica y fisiopatológica actualizada, que deberá ser útil tanto al experto como alno experto en CPC6.
Para el uso en la práctica clínica, se han reconocido cuatro fenotipos diferenciados, que difieren enel manejo a aplicar y en las respuestas al tratamiento (tabla 1). El primer grupo está formado porlos pacientes con SE, en el estadio final de laHAP-CPC, pero en los que se obtiene un efecto beneficioso con el uso de las nuevas terapias emergentes. El segundo grupo engloba a los pacientescon una HAP asociada a cortocircuitos sistémicopulmonares, en los estadios más tempranos de laenfermedad. Estos pacientes con un estado previoal SE presentan un aumento leve o moderado de laRVP. A diferencia de los pacientes con SE, a menudo no se los incluye en los estudios y su tratamiento resulta, por lo tanto, difícil.

El tercer grupo incluye a los pacientes con un cortocircuito cardiaco pequeño que no se cree que seala causa de la HAP. Son muy similares a los pacientes con una HAP idiopática.
El último grupo lo forman los pacientes con unaHAP persistente o recurrente tras una correcciónquirúrgica satisfactoria del defecto cardiaco congénito. Su pronóstico muy negativo resalta la necesidad de unos criterios de operabilidad más exactos.
Estos diversos grupos requieren estrategias demanejo diferentes y presentan distintas respuestasal tratamiento.
Se ha propuesto también una clasificación anatomopatológica-fisiopatológica de los cortocircuitossistémico-pulmonares congénitos asociados a HAP,con objeto de describir mejor el tipo de comunicación congénita (tabla 2).

HISTORIA NATURAL: SÍNDROME DEEISENMENGER
Corrección quirúrgica/operabilidad
Los avances realizados en cirugía cardiaca pediátrica permiten actualmente una cirugía correctora de las CPC asociadas a un aumento del flujosanguíneo pulmonar, que puede aplicarse a unaedad muy temprana. Estas intervenciones van destinadas a prevenir toda una serie de secuelas, entrelas que se encuentra el desarrollo de una HAP y laEVP. Sin embargo, en algunos pacientes con cortocircuitos izquierda-derecha, estos defectos congénitos pueden pasar inadvertidos hasta una faseavanzada de la infancia o hasta la edad adulta y sediagnostican tardíamente, cuando se han producido ya lesiones vasculares pulmonares. En lospaíses en desarrollo, dado que no ha habido antesla oportunidad de cerrar estos defectos en el lactante, la HAP en los niños con CPC es más frecuente. Tal situación está pasando a ser un problema muy real, ahora que empieza a mejorar laasistencia sanitaria en esos países11. En consecuencia, hay una necesidad real de directricessobre la reparación quirúrgica completa o la cirugía paliativa de las CPC en los pacientes que,como consecuencia del trastorno que presentan,sufren un cierto grado de EVP.
¿Cómo puede identificarse con exactitud a los pacientes con un riesgo elevado de HAP persistente tras la cirugía? Los médicos suelen basar su decisiónactualmente en criterios diferentes sobre si un paciente es o no un candidato quirúrgico adecuado.No hay un consenso general y sólo es posible hacerrecomendaciones, en vez de dar unas directrices definitivas12.
La reparación quirúrgica en pacientes con unaRVP elevada y una HAP establecida comportariesgos. Si la RVP continúa siendo alta después dela operación y la HAP persiste, el pronóstico esmalo13. En un estudio retrospectivo de 5 años llevado a cabo en niños con HAP en Reino Unido, lasubpoblación de pacientes con CPC-HAP postoperatoria presentó una evolución mucho peor que lade los pacientes con una HAP asociada a una CPCcompleja (no operada) y SE. Casi una cuarta partede estos niños fallecieron (11/47)13. Los niños con SE tuvieron un tiempo acumulativo de supervivencia superior en 1,3 años, lo cual indica que la reparación quirúrgica no es necesariamente siemprela mejor opción.
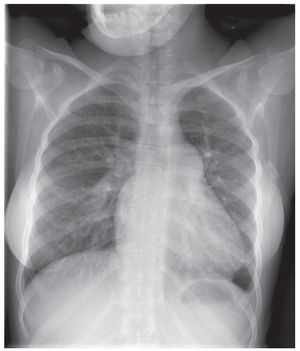
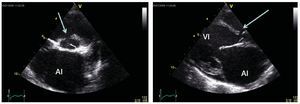
Hay varias exploraciones y criterios que se usanpara informar la decisión de si un paciente con unaHAP asociada a una CPC es un candidato adecuadopara un tratamiento quirúrgico y para determinar elmejor resultado posible que puede alcanzarse: exploración clínica para identificar posibles signos de insuficiencia cardiaca congestiva y saturación de oxígeno, ecocardiografía para detectar signos dehipercirculación pulmonar (figs. 1 y 2) y el métodode referencia actual, que son las determinaciones mediante cateterismo cardiaco derecho de los parámetros hemodinámicos y la vasorreactividad (fig. 3)11,14.

Fig. 1. Radiografía de tórax de un paciente con síndrome de Eisenmengerque presenta una cardiomegalia leve, agrandamiento de las arterias pulmonares y disminución de las marcas vasculares pulmonares periféricas.Estos signos impiden una posible reparación quirúrgica.

Fig. 2. Ecocardiografía de un paciente con una comunicación interventricular y signos de hipercirculación pulmonar que indican una resistencia vascularpulmonar baja y un flujo sanguíneo pulmonar elevado. Estas imágenes suelen señalar como posible la reparación quirúrgica con un cateterismo. En el panelizquierdo se muestra una proyección de eje corto con la comunicación interventricular (flecha) y en particular una aurícula izquierda (AI) dilatada. En el panelderecho se muestra una proyección de eje largo con dilatación del ventrículo izquierdo (VI) y la AI, que indica una hipercirculación pulmonar en presencia deun cortocircuito.

Fig. 3. Cateterismo cardiaco ideal concatéteres que miden simultáneamente lasaturación de oxígeno y las presiones entodas las cavidades y vasos con objetode evitar un sesgo de muestreo temporal.Se describe la fórmula para el cálculo deQp/Qs en los cortocircuitos congénitos,que es el cociente del flujo sanguíneopulmonar (Qp) respecto al flujo sanguíneosistémico (Qs). El gasto cardiaco puedemedirse por termodilución en ausencia decortocircuito o con la fórmula de Fick enpresencia de cortocircuitos intracardiacoso extracardiacos. PAD: presión auricular derecha media; PAI: presión auricularizquierda media; PAP: presión arterialpulmonar media; PCPE: presión capilarpulmonar enclavada. (Cortesía de IngramSchulze Neick National & UK Centre forPulmonary Hypertension in Children,Great Ormond Street Hospital, Londres,Reino Unido.)
Durante un tiempo, se utilizó el examen de las alteraciones histopatológicas de los vasos pulmonaresrealizado mediante una biopsia pulmonar para evaluar la operabilidad15. En la actualidad, la fiabilidadde estos resultados no se considera suficiente para justificar el carácter invasivo y los riesgos asociadosa la obtención de una muestra de tejido16. Además,la biopsia puede no ser representativa de la enfermedad en todo el pulmón, sino tan sólo de unaparte aleatoria. Los pacientes sin un engrosamientode la íntima de las arterias pulmonares —a los que,por lo tanto, se considera que tienen una EVP reversible— pueden contraer de todos modos una HAP postoperatoria irreversible. Además, los niñosde menos edad (< 2 años) con frecuencia son operables a pesar de que la biopsia pulmonar muestre alteraciones avanzadas15. Sin embargo, teniendo encuenta la nueva información que correlaciona losmarcadores de la apoptosis con las alteracionesmorfológicas en el tejido pulmonar y la apariciónde una HAP postoperatoria irreversible9, las biopsias pulmonares continúan desempeñando un papelimportante en la investigación clínica y básica, y deberán seguir haciéndolo. Aún es necesario un mejorconocimiento de la fisiopatología de los vasos sanguíneos pulmonares en la HAP-CPC para poderexaminar plenamente los efectos que ejercen los fármacos y los tratamientos actualmente disponibles ylos que aparezcan en el futuro. Véanse las revisionesdetalladas de Viswanathan et al11, Lopes et al12 yGiglia et al14 sobre los métodos de exploración invasivos y no invasivos para determinar la operabilidad en los pacientes con HAP-CPC.
En la actualidad, los métodos empíricos basadosen los datos hemodinámicos del cateterismo cardiaco derecho y la vasorreactividad se utilizan principalmente para predecir mejor qué pacientes tendrían un resultado quirúrgico positivo o negativo.En un reciente artículo, Lopes et al12, basándose enla literatura médica publicada y en la opinión de expertos de centros de excelencia reconocidos, especificaron unos criterios hemodinámicos basados en laRVP y en el cociente de la resistencia pulmonar respecto a la sistémica, y la forma en que estos valorescambian durante la administración aguda de un vasodilatador. Se determinó que:
1. Un índice de RVP basal < 6 UW/m2 asociadoa un cociente de resistencias < 0,3 sin una pruebade vasorreactividad se interpreta como indicio de una evolución favorable tras las operaciones que establecen una circulación biventricular.
2. Se ha recomendado claramente una prueba deadministración aguda de un vasodilatador, con elempleo de oxígeno/óxido nítrico, si el índice deRVP basal es de 6-9 UW/m2 en presencia de un cociente de resistencias de alrededor de 0,3-0,5.Aunque no hay un consenso absoluto, la operabilidad con un resultado favorable se considera probable si se cumplen los siguientes criterios:
- Una disminución del 20% en el índice de RVP.
- Una disminución de alrededor del 20% en el cociente de la RPV respecto a la sistémica.
- Un índice de RVP final < 6 UW/m2.
- Un cociente de resistencias final < 0,3.
Estas cifras son muy conservadoras y es posibleque tengan que adaptarse en el futuro. La pruebade administrar de forma aguda un vasodilatador,tanto si se trata de óxido nítrico solo como si se usaen una mezcla con oxígeno, es el patrón de referencia como medida de la reactividad del lecho vascular pulmonar17,18. En la HAP idiopática, laspruebas de vasodilatación se realizan para determinar si un paciente responderá al tratamiento conantagonistas del calcio. Sin embargo, cuando setrata de valorar la operabilidad de un paciente conuna CPC y una RVP elevada, no está claro si son lobastante exactas para poder diferenciar por completo entre los pacientes que obtendrán un buen resultado de la cirugía y los que no. Además, hay dificultades técnicas que llevan a errores de cálculo, yes preciso tener presentes otros trastornos médicosal llevar a cabo pruebas con vasodilatadores11. Continúa sin estar claro qué parámetro de la hemodinámica pulmonar preoperatoria presenta una mejor correlación con el resultado de la cirugía. No se conoce por completo de qué forma los factores decada paciente individual, como el tipo de lesión cardiaca o la predisposición genética, influyen en losresultados.
Debe señalarse que los criterios antes citados noson aplicables a los pacientes con una fisiología deventrículo único en los que se está evaluando laconveniencia de crear una circulación de Fontan.En estos casos, lo ideal es que tengan un grado deRVP casi normal y ciertamente no > 3 UW/m2.
Además, la obtención de unas determinaciones hemodinámicas exactas puede resultar aún más difícilen los pacientes con una fisiología de ventrículoúnico14.
Aunque las determinaciones hemodinámicas basadas en el cateterismo cardiaco derecho resultanútiles y, de hecho, son los mejores instrumentos deque disponemos en la actualidad, no están exentas defallos. Los pacientes que se encuentran dentro de loslímites que se considera apropiados para determinarun buen resultado operatorio pueden presentar, detodos modos, una HAP postoperatoria persistente.Son necesarios instrumentos de evaluación mejores,más exactos y, preferiblemente, menos invasivos, sobre todo en los pacientes que se considera que están en una situación limítrofe en cuanto a la operabilidad, dado su perfil hemodinámico. Recientemente, Lévy et al han realizado algunos avances prometedores para desarrollar nuevos instrumentos quepermitan evaluar la operabilidad19-21.
Los pacientes con una hemodinámica similarantes de la operación se estratificaron en dos grupospostoperatoriamente, en función de si tenían o nohipertensión postoperatoria persistente. En lasbiopsias pulmonares, aunque los aumentos delgrosor de la pared arterial pulmonar se apreciaron en todos los pacientes, 10 de los 11 que tenían hipertensión pulmonar irreversible presentaban unengrosamiento pronunciado de la íntima. Esto seacompañaba de la expresión exclusiva del Bcl-2, unmarcador antiapoptótico, por parte de las célulasendoteliales de las arterias con una fibrosis grave dela íntima. No hubo diferencias entre la expresión delos marcadores de la apoptosis caspasa-3 y p53 enlas células endoteliales de los dos grupos. Estosdatos indican que la proliferación de células endoteliales resistentes a la apoptosis puede ser un factorcausal en el engrosamiento de la íntima. La evidencia experimental obtenida en modelos animalesde la HAP respalda la hipótesis de que un factordesencadenante que lleve a la apoptosis inicial delas células endoteliales podría fomentar que despuéssurjan y proliferen células endoteliales resistentes ala apoptosis.
Reconociendo el hecho de que la biopsia depulmón es un método invasivo que no es ideal parala práctica clínica general, Smadja et al intentarondeterminar si las células endoteliales circulantes(CEC) (ya reconocidas como un marcador no invasivo de la lesión, el remodelado y la disfunción vasculares) podrían ser un biomarcador apropiado quepermitiera identificar a los pacientes con un riesgoelevado de sufrir una HAP irreversible tras la reparación de una CPC. Los pacientes con HAP irreversible, además de presentar un engrosamiento de laíntima arterial pulmonar y la correspondiente expresión elevada de Blc-2 en las células endotelialesde la biopsia de pulmón, mostraban también cantidades de CEC en sangre periférica significativamente superiores que los pacientes con una HAPreversible20. A diferencia de las CEC, otros biomarcadores de la activación, la regeneración y la lesióndel endotelio no han permitido diferenciar la HAPreversible de la irreversible tras la cirugía.
Pre-Eisenmenger no operable
Como se ha mencionado antes, el segundo grupode la subclasificación incluye a los pacientes conuna RVP que se considera demasiado alta para unareparación quirúrgica, pero que no llegan al diagnóstico de SE. Hasta el momento, el enfoque terapéutico utilizado para este grupo de pacientes hasido el de observación y vigilancia a la espera deque se desarrolle un SE. Sin embargo, con la aparición de las nuevas terapias dirigidas utilizadas paratratar la hipertensión pulmonar en diversos contextos, ha surgido el concepto de tratar y reparar,¡con más preguntas que respuestas!22.
En los últimos 10 años, los análogos de las prostaciclinas, los antagonistas de los receptores de la endotelina y los inhibidores de la fosfodiesterasatipo 5 (PDE-5) han resultado eficaces en el tratamiento de la HAP, en parte a través de acciones vasodilatadoras23. Se ha propuesto que los antagonistas de los receptores de la endotelina enparticular pueden tener otras acciones adicionales,como la prevención del crecimiento y la fibrosis delas células endoteliales, y que ejercen un efecto deremodelado en el lecho vascular pulmonar. Dehecho, sus acciones vasodilatadoras son de menorinterés en la discusión de la posible capacidad depreparar a los pacientes con HAP-CPC para la operabilidad; son sus acciones antiproliferativas y elpotencial de inducir una regresión de las lesiones loque tiene mayor importancia.
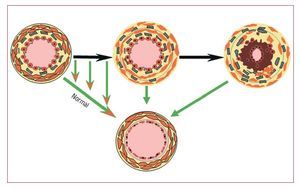
El antagonista dual de los receptores de la endotelina bosentán se ha investigado ampliamente en laHAP idiopática y hay también una evidencia sólidaque indica su eficacia en el tratamiento de la HAPasociada a las CPC24. A diferencia de lo que ocurreen la HAP idiopática, la causa de la HAP en los pacientes con CPC que presentan defectos grandes esen parte conocida; se cree que la carga de presión yde volumen ejercida contra el lecho vascular pulmonar conduce a un remodelado y lesiones vasculares pulmonares. Mientras que los pacientes conuna HAP idiopática presentan siempre lesiones definidas, no siempre sucede así en el caso de los pacientes con CPC, en quienes las lesiones pueden sermenos amplias. Así pues, mediante la reducción dela RVP en los pacientes en que las lesiones vasculares no son amplias, surge la posibilidad de utilizarun pretratamiento con vasodilatadores para mejorar el estado de un paciente, con lo que un casoinoperable podría considerarse operable (fig. 4). Sinembargo, es posible que no siempre sea así en lospacientes con lesiones amplias y EVP establecida.Aunque los fármacos puedan reducir la RVP enestos pacientes, la HAP podría persistir postoperatoriamente, y ello podría comportar un peor pronóstico.

Fig. 4. Concepto «tratar y reparar». En laparte superior se muestra la progresión delas lesiones en presencia de un cortocircuito izquierda-derecha. La flecha indicala oportunidad de observar una regresiónde las lesiones a un estado normal o casinormal. Esto brinda la oportunidad de tratar a los pacientes con un aumento de laresistencia vascular pulmonar que contraindica la cirugía, con objeto de remodelarel lecho vascular y hacer posible una corrección completa de la lesión anatómicasubyacente.
Un posible problema que puede aparecer al reducir la RVP con terapias dirigidas a la HAP es queun aumento del flujo sanguíneo pulmonar, debido aun incremento de la presión en la comunicacióncardiaca, restablezca la propensión a la apariciónde la lesión. Así pues, paradójicamente, la reversióndel remodelado vascular y la formación de la lesión,al dar lugar a un descenso inicial de la RVP, dehecho podrían producir después una lesión vascularpulmonar. Una solución sería aplicar una banda enla arteria pulmonar una vez se ha reducido la RVP,con objeto de reducir el flujo sanguíneo al lechovascular pulmonar y evitar una mayor lesión.
Además de las acciones vasodilatadoras, el bosentán tiene también acciones antifibróticas, antiproliferativas y antiinflamatorias. Los análogos de prostaciclina inhiben la agregación plaquetaria y elcrecimiento de las células de músculo liso. Las propiedades adicionales de estos fármacos pueden desempeñar también un papel en cuanto a prevenir o retardar el remodelado vascular. Se han descrito varioscasos de pretratamiento con prostaciclinas o bosentán utilizados antes de la cirugía de las CPC, conobjeto de preparar a pacientes en situación limítrofeo «inoperables»25-28. Estos casos indican una ventajadel empleo de análogos de las prostaciclinas o antagonistas de los receptores de la endotelina para mejorar la hemodinámica y crear unas condiciones másfavorables para la reparación. Sin embargo, hay varios elementos muy importantes que es preciso teneren cuenta. La mayor parte de los pacientes presentaban comunicaciones interauriculares (CIA) simples. La evaluación de la operabilidad puede sercuestionable. En la mayor parte de los casos, eltiempo de seguimiento de los pacientes cuando serealizó la presentación de los resultados era corto(1 año o menos). Para poder establecer con seguridad un resultado satisfactorio, serán necesariosdatos correspondientes a un mínimo de varios años.Aunque estos casos aislados muestran un éxito delpretratamiento previo a la operabilidad, no sabemosen cuántos casos esto ha fracasado y no se ha notificado. Sería necesario un análisis retrospectivo de losregistros nacionales sobre este tipo de datos parapoder disponer de una imagen completa.
HIPERTENSIÓN PULMONAR PERSISTENTETARDÍA TRAS LA REPARACIÓN QUIRÚRGICA
El estado funcional y estructural del lecho vascularpulmonar desempeña un papel clave en la forma depresentación y en la evolución de los niños con enfermedades cardiovasculares congénitas. Es el periodopostoperatorio inmediato cuando el niño presenta la máxima vulnerabilidad a un aumento súbito o sostenido de la RVP. Tras la cirugía de una CPC, la reactividad vascular pulmonar aumenta y los estímulosvasospásticos pueden comportar un aumento súbitode la PAP y la resistencia, que dará lugar a una insuficiencia cardiaca derecha aguda, insuficiencia tricuspídea, hipotensión sistémica, isquemia miocárdica yaumento de la resistencia de las vías aéreas29. Estoseventos, denominados crisis hipertensivas pulmonares, pueden ser mortales. Eventos levemente estimulantes pueden desencadenar crisis similares, y dichas crisis tienden a tener una mayor duración yaparecer de forma agrupada.
Sin embargo, con la mejora de la asistenciapostoperatoria y la introducción de nuevas terapias,las crisis de hipertensión pulmonar aguda puedentratarse en la mayor parte de los casos.
La incidencia de episodios de hipertensión pulmonar postoperatoria se redujo del 31% en el periodo comprendido entre 1980 y 1984 al 6,8% antesdel uso sistemático de óxido nítrico inhalado30. Lasseries que reflejan la práctica clínica contemporáneaindican que la hipertensión pulmonar aparece comocomplicación en el 2% de los pacientes a los que sepractican intervenciones quirúrgicas para CPC, yque se producen crisis en un 0,75%7. Sin embargo,la mortalidad en los pacientes que sufren una crisiscontinúa siendo alta, de un 20%, y se identifica unaVP como factor contribuyente importante en la duración de la hospitalización y la necesidad de ventilación mecánica prolongada. (Para mayor información, véase la revisión reciente de Adatia et al29, yaque esta cuestión no entra en lo tratado en el presente artículo.)
No obstante, el tratamiento agudo y la supervivencia en el periodo postoperatorio inmediato no implican que la hipertensión pulmonar se resuelva,y el paciente puede presentar una HAP persistentetras la reparación quirúrgica, pero sin que haya cortocircuito. Es poco lo que se sabe acerca de estegrupo específico de pacientes, puesto que la literatura al respecto es escasa. A este grupo se lo ha incluido en la subclasificación de la CPC-HAP(grupo 4 de la subclasificación clínica)6. Las características hemodinámicas que presenta son muy similares a las de la HAP idiopática y, por lo tanto, elpronóstico parece malo según lo indicado por losdatos recientes13. Esto subraya la importancia deuna decisión exacta, por cuanto la operabilidad y lasupervivencia podrían ser incluso mejores con uncortocircuito abierto y un SE que con un cortocircuito cerrado y una insuficiencia ventricular derecha31.
TRATAMIENTO DEL SÍNDROMEDE EISENMENGER
El SE es la forma más avanzada de HAP asociada a las CPC. Los signos y síntomas de SE suelendeberse a una baja saturación de oxígeno de lasangre y consisten en disnea, cianosis, fatiga,mareo, síncope y arritmias. Los síntomas puedenno aparecer hasta una fase avanzada de la infanciao el inicio de la edad adulta. En general, los pacientes con SE presentan una esperanza de vida reducida, aunque pueden sobrevivir hasta la tercera ola cuarta década de la vida32, y algunos de ellosllegan incluso a la séptima década con un tratamiento apropiado32,33. De todos los pacientes conCPC, los que presentan un SE son los que muestranun deterioro más grave en cuanto a intolerancia alejercicio34. Se ha identificado que tal intolerancia alejercicio en este tipo de pacientes es un predictor dehospitalización o muerte independientemente de laedad, el sexo, la clase funcional o el defecto cardiaco subyacente34. Hay evidencia testimonial deque los pacientes con SE adaptan su estilo de vidaen función de su capacidad de ejercicio y tienden asubestimar sus limitaciones. A pesar de ello, el SEafecta de forma clara y grave a la capacidad de ejercicio del paciente, por lo que reduce su calidad devida.
Tratamiento convencional
En publicaciones recientes se ha descrito ampliamente el tratamiento del SE2,5,35. Como se ha mencionado antes, las opciones de tratamiento en lospacientes con SE se han limitado históricamente amedidas paliativas y el trasplante de corazón ypulmón (TCP). El tratamiento se ha basado lamayor parte de las veces en el uso de digitálicos,diuréticos, antiarrítmicos y/o anticoagulantes. Sin embargo, ninguna de estas clases de medicamentosmodifica la supervivencia ni influye de forma significativa en el riesgo de deterioro clínico en el SE. Seha utilizado digoxina en el tratamiento paliativo dela insuficiencia cardiaca derecha en el SE, aunque laevidencia existente para respaldar su empleo es especialmente débil36. El uso de anticoagulación enpacientes con SE es motivo de controversia, puestoque estos pacientes presentan una incidencia elevada de trombosis arterial pulmonar, hemoptisis eictus y aumento del riesgo de hemorragia36. En unestudio reciente se ha estimado que la prevalenciade la trombosis arterial pulmonar en el SE es del20%, con un riesgo que se correlaciona con el aumento de la edad, la disfunción biventricular, la dilatación de las arterias pulmonares y la reducciónconcomitante de la velocidad del flujo pulmonar37. Aunque la evidencia existente indica un efecto beneficioso de este tratamiento en los pacientes conHAP idiopática, no hay datos en el SE, y los riesgosasociados de hemorragia en el tratamiento de estospacientes pueden ser superiores a los posiblesefectos beneficiosos. Hasta la fecha, no se han realizado estudios prospectivos en los que se haya abordado la utilidad de la anticoagulación en la prevención de la trombosis o la hemoptisis, y hay una grannecesidad de datos de este tipo38.
La eficacia de los antagonistas del calcio en lospacientes con SE no está demostrada ni se los recomienda de manera general, puesto que su uso puedecausar una disminución aguda de la presión arterialsistémica y un aumento del cortocircuito derechaizquierda que puede conducir a síncopes y muertesúbita39. La oxigenoterapia domiciliaria a largoplazo, durante un mínimo de 12-15 h diarias, puedemejorar los síntomas, pero no se ha demostradoque modifique la supervivencia36.
La educación sanitaria del paciente, las modificaciones de la conducta y el conocimiento de los posibles factores de riesgo médicos son aspectos importantes del manejo de estos pacientes. Los pacientescon SE presentan un especial riesgo durante lasoperaciones de cirugía cardiaca o no cardiaca y laanestesia, así como por las consecuencias de la deshidratación, las infecciones respiratorias, la gran altitud y las vías intravenosas. También se recomienda que eviten el ejercicio extenuante y losdeportes de competición.
El embarazo se asocia a un riesgo elevado, tantopara la madre como para el feto. Las tasas deaborto espontáneo son altas y tan sólo alrededor deun 25% de los embarazos llegan a término. De losrecién nacidos que sobreviven, aproximadamenteuna tercera parte presenta signos de retraso del crecimiento intrauterino y la mortalidad perinatal eselevada. La mortalidad materna es de aproximadamente un 45% entre las pacientes con SE, y la muerte suele producirse durante el parto o en la primera semana siguiente, en la mayor parte de loscasos como resultado de tromboembolias, hipovolemia o preeclampsia. Así pues, el embarazo está contraindicado en las pacientes con SE40,41.
Terapias dirigidas
Como se ha comentado antes, parece claro que elsistema de la endotelina 1 desempeña un papel importante en las anomalías estructurales y funcionales que se producen en los vasos sanguíneos pulmonares y en la progresión de la HAP en todas lasformas del trastorno, incluida la HAP-CPC. Dadoque el tratamiento con antagonistas de los receptores de la endotelina ha sido eficaz en los pacientescon HAP idiopática o asociada a enfermedades deltejido conjuntivo (HAP-ETC)42-44, sería de esperarque tuvieran unos efectos beneficiosos similares enlos pacientes con HAP-CPC. El primer ensayo clínico aleatorizado, a doble ciego y controlado conplacebo en pacientes con SE fue el BREATHE-5(Bosentan Randomised Trial of Endothelin Antagonist-5), en el que se investigó la eficacia del antagonista dual de los receptores de la endotelina bosentán en 54 pacientes adultos con SE. Durante esteestudio de 16 semanas, el bosentán redujo significativamente la RVP y la PAP media y mejoró la capacidad de ejercicio en comparación con el grupo placebo, sin influir negativamente en la saturación deoxígeno arterial sistémica45. Este resultado de seguridad es de especial importancia en los pacientes conSE, dada la posibilidad de que se agrave el cortocircuito en general por la posible reducción de la resistencia sistémica en respuesta a los tratamientos vasodilatadores. Los datos de seguimiento más largo,en el estudio de extensión abierto de los pacientesque participaron en el ensayo a doble ciego inicialde 16 semanas, mostraron que las mejoras de la capacidad de ejercicio se mantenían otras 24 semanasde tratamiento46. La clase funcional mejoró tambiénen ese lapso, y el tratamiento fue bien tolerado.
Estos resultados están respaldados por diversosestudios abiertos, a pequeña escala, que muestrantambién una mejora de la clase funcional, la saturación de oxígeno, el estado clínico y la hemodinámica pulmonar en pacientes pediátricos y adultoscon SE47-50. Los datos obtenidos a largo plazo indican que las mejoras se mantienen durante 2 añosde tratamiento, sin que surjan problemas de seguridad o tolerabilidad51,52, pero otros estudios han indicado que el efecto puede disminuir con el paso deltiempo53. Los resultados de estos estudios ponen enduda el dogma de que la EVP en pacientes con SE no responde al tratamiento. Por otro lado, el SE no es una enfermedad estable como se había supuesto, sino que presenta un deterioro progresivo, como indica el aumento de la RVP que se observó en lospacientes del grupo placebo del estudio BREATHE-545.
Los datos del estudio BREATHE-5 señalan también que el lugar en que se encuentra el defecto deltabique no influye en la hemodinámica a corto plazoni en las mejoras funcionales obtenidas con el tratamiento de bosentán. Dado que la evolución de la enfermedad vascular pulmonar en los pacientes conSE que presentan una CIA difiere notablemente dela de los pacientes con una comunicación interventricular (CIV), su respuesta al tratamiento médicopodría ser diferente también. En un análisis post-hoc del ensayo BREATHE-5, se compararon los efectosdel bosentán y de placebo en pacientes con CIA y enpacientes con CIV o con ambos defectos (CIV/CIA+CIV)54. En ambos subgrupos, se observó ausencia de cambios de la oximetría de pulso sistémicaentre ambos grupos de tratamiento, y los efectos deltratamiento, corregidos para el efecto placebo, laRVP indexada, la capacidad de ejercicio y la PAPmedia, también fueron comparables.
Estamos todavía a la espera de datos sobre otrosantagonistas de los receptores de la endotelina en laHAP-CPC, pero sería de esperar que los resultadosfueran similares a los obtenidos en otras formas deHAP.
Inhibidores de la fosfodiesterasa tipo 5
Hasta la fecha, los datos existentes sobre el usode los inhibidores de PDE-5 en pacientes con SEson escasos. Después de 6 meses de tratamiento, laclase funcional de la Organización Mundial de laSalud (OMS), la saturación de oxígeno y la PAPmedia y sistólica y la RVP presentaron una mejorasignificativa en 7 pacientes con SE que participaronen un ensayo prospectivo y abierto de sildenafilopero, aunque hubo una tendencia a la mejora, loscambios de la distancia recorrida en 6 min no alcanzaron significación estadística55. Hubo pocos efectossecundarios significativos y, aunque existía la posibilidad teórica de una reducción del flujo sanguíneopulmonar como consecuencia de la disminución dela resistencia vascular sistémica, de hecho la cianosis mostró una mejora en estos pacientes. Laclase funcional, la capacidad de ejercicio y la hemodinámica pulmonar presentaron también una mejora, sin efectos secundarios significativos en 21 pacientes con SE tratados con sildenafilo, en unensayo prospectivo de dosis-respuesta, no aleatorizado y no controlado56.
Estos resultados están respaldados por los deotros estudios pequeños de los inhibidores dePDE-5, solos o en combinación con prostanoides,que han mostrado también una mejora de la capacidad de ejercicio, la clase funcional y algunos parámetros hemodinámicos, sin que hubiera problemasde seguridad57-60. Recientemente, Tay et al demostraron que 3 meses de sildenafilo eran bien tolerados por 12 adultos con SE, y que su uso se asociaba a una mejora significativa de la calidad devida y la capacidad de ejercicio61. Los resultados relativos a la eficacia, aunque alentadores, deberánser validados en amplios ensayos aleatorizados ycontrolados con placebo. En la actualidad se estáreclutando a pacientes en Alemania para un ensayode este tipo (para investigar los efectos del sildenafilo en la capacidad de ejercicio y la hemodinámicacardiopulmonar en pacientes con SE). Además,también están por determinar los efectos y la efectividad a largo plazo en pacientes con SE que presentan defectos subyacentes más complejos.
Prostaciclina y análogos de la prostaciclina
En general hay pocos datos y no disponemos deningún ensayo amplio sobre el uso de prostanoidesen el SE. El tratamiento con prostaciclina intravenosa a largo plazo mejoró la hemodinámica y laclase funcional en 20 pacientes con HAP asociada adiversos tipos de CPC, aunque ninguno de los pacientes presentó una respuesta hemodinámicaaguda62. La administración intravenosa continua deepoprostenol produjo una mejoría significativa dela clase funcional, la saturación arterial y la distancia recorrida en 6 min, y redujo la RVP en 8 pacientes con SE después de 3 meses de tratamiento63. Sin embargo, en una serie anterior, el tratamientode los pacientes con epoprostenol había producidoacontecimientos adversos, como aumento de la resistencia vascular sistémica, aumento de la RVP yreducción del oxígeno arterial en 8 de 10 pacientes64. Además, se han registrado diversos acontecimientosadversos, como accidentes cerebrovasculares, queprobablemente se debieron al uso de un catéter venoso central en presencia de un cortocircuito derecha-izquierda63. Dada la mediana de supervivencia,más larga en los pacientes con SE que en los pacientes con una HAP idiopática, los posibles riesgosde usar catéteres a largo plazo son especialmenteimportantes al analizar la relación riesgo-beneficiode un tratamiento. Los datos existentes sobre otrasformas de terapia con prostanoides, incluidos el iloprost inhalado o intravenoso y el beraprost oral enel SE, se limitan a presentaciones de casos, estudiosde casos y series pequeñas. Los prostanoides inhalados y orales aportan ventajas evidentes respectoal epoprostenol en cuanto a seguridad de la administración a largo plazo, pero su eficacia y su seguridad no se han estudiado todavía de manera completa en esta población de pacientes.
En una reciente publicación de Dimopoulos et alse ha evidenciado una mejora de la supervivencia en el SE con el empleo de diferentes tipos de terapiasdirigidas, y ello ha confirmado el potencial queaporta esta forma de abordar el SE65.
Trasplante de órganos torácicos
En última instancia, el trasplante, preferiblementeel TCP, constituye una opción de tratamiento tansólo en un subgrupo pequeño de pacientes seleccionados, y tiene importantes limitaciones derivadasde la disponibilidad de órganos de donantes. Eléxito del trasplante varía en función de cuál sea lacausa subyacente del SE, y su efecto más favorableparece ser el que se obtiene en los pacientes conCIV o con anomalías congénitas múltiples66. En general, el trasplante en pacientes con SE se asocia auna mortalidad perioperatoria elevada67. Sin embargo, los estudios realizados indican que, aunqueel curso postoperatorio tienda a ser complicado enestos pacientes, las tasas de supervivencia a corto ya largo plazo tras un TCP son similares a las descritas en pacientes trasplantados que no presentanun SE68. Se han alcanzado tasas de supervivencia a1 año de aproximadamente un 70% tras el TCP, ydel 55% tras el trasplante de pulmón. Las tasas desupervivencia a 5 y 10 años tras un TCP son del 51y el 28% respectivamente66,68.
Dados la escasez de órganos donantes apropiados, el bajo número de receptores apropiados yel mal pronóstico tras el TCP, toda medida que permita retrasar la necesidad del TCP en los pacientescon SE será muy bien recibida. Un análisis retrospectivo reciente señala que los pacientes con SE querecibieron los nuevos tratamientos avanzados,como los análogos de la prostaciclina o los antagonistas de los receptores de la endotelina, puedenhaber alcanzado un aumento del tiempo mediotranscurrido hasta la muerte o la inclusión en lalista de espera activa, en comparación con los pacientes que no recibieron dichos tratamientos69. Dada la falta de efectos beneficiosos con el uso deltratamiento convencional en el SE, y teniendo encuenta las escasas opciones quirúrgicas disponiblesuna vez instaurada la enfermedad, hay una claranecesidad médica no satisfecha en pacientes con SEque puede abordarse con las terapias dirigidas.
La circulación de Fontan
Desde que se describiera por primera vez hacemás de tres décadas, la operación de Fontan y susvariaciones han pasado a ser las intervenciones deelección para el tratamiento de los pacientes concardiopatías congénitas que presentan un ventrículoúnico anatómico o funcional. El objetivo de la operación de Fontan es utilizar este ventrículo únicopara impulsar la circulación sistémica, al tiempo que la circulación pulmonar es impulsada principalmente por la presión intratorácica negativa.
En la actualidad no hay ningún tratamiento médico general satisfactorio para los casos de fracasode la circulación de Fontan. El tratamiento ha consistido en abordar las manifestaciones específicas,como la disfunción ventricular, la enteropatía conpérdida de proteínas y el aumento de la RVP. En los pacientes a los que se ha practicado una operación de Fontan se ha intentado el tratamiento de ladisfunción ventricular con el empleo de diversosagentes, pero hay pocos datos que lo respalden y laevidencia existente indica que generalmente aportanun beneficio escaso o nulo, debido a su falta de influencia en la reducción de la precarga70. Los inhibidores de la enzima de conversión de la angiotensinano tienen efecto alguno en la capacidad de ejercicio,la resistencia vascular sistémica, el índice cardiacoen reposo o la función diastólica en los pacientescon circulación de Fontan71. No obstante, a pesarde esta falta de evidencia, muchos pacientes con circulación de Fontan de larga evolución reciben tratamiento.
El bajo gasto cardiaco, la hipoxemia excesiva o laenteropatía con pérdida de proteínas pueden sermanifestaciones clínicas de un aumento de la RVP;por consiguiente, su prevención o su tratamientopueden ser de crucial importancia. En la fase postoperatoria aguda, los pacientes con un aumento dela RVP son tratados con óxido nítrico y oxígenoadicional. El óxido nítrico inhalado reduce la presión venosa central72, la PAP media y el gradientede presión transpulmonar. Las prostaciclinas se hanutilizado con poca frecuencia en el periodo perioperatorio en pacientes con intervención de Fontan, yhay pocos datos al respecto. El beraprost reduce laPAP media y la RVP en la fase preoperatoria en pacientes candidatos a la operación de Fontan quepresentan una hipertensión pulmonar leve73, y se hademostrado que el epoprostenol previene el efectorebote tras el cese de la administración de óxido nítrico inhalado en la fase postoperatoria inicial74. Cuando empieza a ser posible la administraciónoral, en el periodo postoperatorio se utiliza con frecuencia sildenafilo, un inhibidor de la PDE-5 queactúa como vasodilatador, puesto que se consideraseguro y tiene un inicio de acción rápido y una altaefectividad. Sin embargo, el sildenafilo no ha sidoautorizado para este uso y no hay datos publicadosque respalden su eficacia o su seguridad en esta indicación.
En el tratamiento de los pacientes con un fallo dela circulación de Fontan, se han publicado hasta lafecha pocas observaciones que describan los efectosde los fármacos que reducen la RVP. El tratamientode los pacientes con circulación de Fontan en unafase tardía con óxido nítrico inhalado reduce la RVP, aunque carece de efectos significativos en elíndice cardiaco75. La vasodilatación pulmonar dependiente de NO y mediada por guanosinmonofosfato cíclico (GMPc) puede potenciarse también conel empleo de sildenafilo. Se ha demostrado que unadosis única de sildenafilo mejora la capacidad deejercicio y la respuesta hemodinámica al ejercicio delos pacientes con circulación de Fontan en una faseavanzada y sin fallo funcional76. También hay estudios de casos aislados que muestran mejoría en unpaciente con una bronquitis plástica y en un paciente con enteropatía con pérdida de proteínas trasla administración de sildenafilo77,78. El efecto del sildenafilo en la tolerancia al ejercicio, la función ventricular y la calidad de vida se está estudiando actualmente en niños a los que se ha practicado unaoperación de Fontan. En la actualidad no disponemos de datos sobre la efectividad del tratamientocon prostanoides en pacientes con un fallo de la circulación de Fontan.
Se ha demostrado que el antagonista dual de losreceptores de la endotelina bosentán mejora la capacidad de ejercicio, la clase funcional, la calidad devida y los parámetros hemodinámicos, incluidas laRVP y la PAP, en pacientes con HAP. El tratamiento a largo plazo con bosentán produjo una mejoría sintomática y de la saturación de oxígeno aórtica, la clase funcional de la OMS, la capacidad deejercicio máxima y submáxima, el índice de disneade Borg, la PAP media, el flujo sanguíneo pulmonary la RVP en un paciente con una bronquitis plásticatras una operación de Fontan79. En un pequeño estudio reciente, la saturación de oxígeno mejoró en5/9 pacientes durante un periodo de tratamiento de16 semanas con bosentán80. Los antagonistas simples de los receptores de la endotelina ambrisentány sitaxentán se utilizan también en el tratamiento dela HAP; sin embargo, no disponemos de datos quedescriban su uso en pacientes con circulación deFontan.
Dados el importante papel que desempeña la circulación vascular pulmonar en la fisiología deFontan, el aumento demostrado de la PAP con laedad y la actual falta de datos, hay una clara necesidad de estudios clínicos sobre la eficacia y la seguridad de los posibles tratamientos para los pacientescon un fallo de la circulación de Fontan, en los quepuedan fundamentarse las tan necesarias recomendaciones terapéuticas.
CONCLUSIONES
En resumen, las mejoras alcanzadas en el diagnóstico y el tratamiento médico y quirúrgico hanmodificado las perspectivas de supervivencia alargo plazo en los pacientes con HAP-CPC, y elloha dado lugar a un aumento significativo del número de pacientes que sobreviven hasta la edadadulta. Aunque existen algunas diferencias en laetiología, la respuesta al tratamiento y la supervivencia, los pacientes pediátricos y adultos conHAP-CPC tienen básicamente la misma enfermedad; un trastorno complejo con una historia natural que varía en función del defecto cardiaco subyacente y de la adaptación crónica multiorgánica aeste. Los efectos beneficiosos a corto plazo de lasnuevas terapias dirigidas en la HAP-CPC son cadavez más claros, pero será necesario el estudio alargo plazo en poblaciones con HAP-CPC. Tieneun interés creciente el tratamiento de los pacientescon defectos congénitos complejos, así como el momento adecuado para la intervención y la operabilidad de los pacientes para optimizar los resultados;cada una de estas cuestiones requiere más estudio.Concretamente, serán necesarios nuevos estudiossobre el papel de las terapias dirigidas y el posibleconcepto de tratar y reparar. Otro reto que se debeafrontar es la fisiología de Fontan que no cumplelos criterios de diagnóstico de la HAP pero en laque desempeña un papel central la circulación pulmonar. A pesar de los avances que se han producido, continúa habiendo mucho trabajo por hacerpara alcanzar un mejor conocimiento de este problema que permita abordar mejor la HAP en esapoblación.
ABREVIATURAS
CEC: células endoteliales circulantes.
CPC: cardiopatía congénita.
EVP: enfermedad vascular pulmonar.
HAP: hipertensión arterial pulmonar.
PAP: presión arterial pulmonar.
RVP: resistencia vascular pulmonar.
SE: síndrome de Eisenmenger.
TCP: trasplante de corazón y pulmón.
Full English text available from: www.revespcardiol.org
Conflictos de intereses:
El Profesor Maurice Beghetti ha formado parte de consejos asesores oha sido consultor de Pfizer, Actelion Pharmaceuticals, Bayer Schering,GlaxoSmithKline, INO therapeutics, Eli Lilly y Mondobiotech y ha recibidopagos por conferencias de Actelion Pharmaceuticals, Encysive, Pfizer yBayer Schering.
Correspondencia: Prof. M. Beghetti.
Pediatric Cardiology Unit. Department of the Child and Adolescent.Children's Hospital. University of Geneva.
6 rue Willy-Donzé. CH-1211 Geneva 14. Suiza.
Correo electrónico: Maurice.Beghetti@hcuge.ch