Palabras clave
Desde que la displasia arritmogénica del ventrículo derecho fue descrita por primera vez por Dalla Volta et al1 en 1961, y posteriormente caracterizada por Fontaine et al2 en 1977, hasta la actualidad, en la que ha sido incluida en la clasificación de la Organización Mundial de la Salud de las miocardiopatías3, las aportaciones a la literatura acerca de esta enfermedad han sido numerosas4,5. Inicialmente las descripciones se centraban en el sustrato arrítmico de ciertas zonas del ventrículo derecho (VD), el llamado «triángulo de la displasia», pero actualmente el espectro se ha ampliado para dar paso a manifestaciones difusas en el VD, a la afección única ventricular izquierda y biventricular en fase dilatada, a menudo indistinguible de la miocardiopatía dilatada (MCD)6.
GENÉTICA Y PATOGÉNESIS
La agregación familiar se demuestra hasta en el 50% de los casos. El tipo de herencia es autosómico dominante con expresión variable y penetrancia incompleta (30%)7. La forma autosómica recesiva también se ha descrito, asociada a la enfermedad de Naxos8,9 y a mutaciones en el gen codificador de la desmoplaquina (DSP)10.
Se han descrito 9 loci asociados con la miocardiopatía arritmogénica del VD (MAVD). Y en la actualidad existen mutaciones en 3 genes relacionadas causalmente con la enfermedad. La tabla 1 resume los loci aislados mediante técnicas de mapeo cromosómico.

Aunque la causa de la MAVD es aún desconocida, se barajan distintas teorías. En la teoría inflamatoria, apoyada por la aparición de infiltrados inflamatorios en series necrópsicas, el daño miocárdico vendría explicado por un proceso continuado de daño y reparación simulando una miocarditis crónica. En el diagnóstico diferencial de la MAVD se deben incluir la miocarditis crónica con afección única del VD, lo que aumenta la complejidad a la hora de reflexionar respecto de su etiología. En la teoría genética, las mutaciones en genes que codifican proteínas específicas darían lugar a la «distrofia» miocárdica. En este sentido, los recientes descubrimientos de mutaciones causales de la enfermedad inducen a esbozar nuevas teorías patogenéticas basadas en el estrés mecánico intercelular. Los estudios descriptivos señalan que la sustitución progresiva del miocardio por células del tejido adiposo y del tejido fibroso sucede tras un exagerado e inadecuado proceso de apoptosis11-13. Los modelos animales apoyan el desequilibrio provocado por el estrés mecánico intercelular como un desencadenante de la apoptosis14,15.
Alteraciones de las uniones intracelulares
Uno de los potenciales mecanismos del origen de la MAVD es la pérdida progresiva de miocitos secundaria a alteraciones estructurales. Este mismo mecanismo se ha descrito en la génesis de la MCD16.
Las uniones intercelulares están constituidas por proteínas; entre ellas, la placoglobina (JUP) y la DSP desempeñan un papel clave en la transducción del estrés mecánico y la comunicación intracelular17. Ambas se encuentran presentes tanto en los miocitos cardíacos como en las uniones epidérmicas18,19. La JUP es una proteína citoplasmática que forma parte tanto de los desmosomas20 como de las uniones adherentes. Participa en la unión de los filamentos intermedios y el citoesqueleto de la actina con los complejos transmembrana que conectan las células adyacentes. La forma mutada de la JUP21 favorece un sustrato intercelular inestable9. La JUP, además, regula la expresión de la proteína antiapoptótica BCL-222.
La DSP es un componente de la placa desmosómica, ancla los filamentos intermedios a la membrana plasmática y constituye una plataforma esencial para el mantenimiento de la integridad celular. En las células de Purkinje también sirve de unión para la desmina23.
Aunque la primera mutación del gen que codifica la JUP está relacionada con la transmisión de la forma recesiva en la enfermedad de Naxos (queratoderma palmoplantar no queratolítico, cabello lanoso y MAVD), descrito en el brazo largo del cromosoma 178,24, el descubrimiento de otras mutaciones no ha hecho más que empezar. Una mutación S299R en el exón 7 de la DSP se ha descrito en una familia con herencia autosómica dominante con MAVD25. La tríada alteraciones cutáneas, pelo lanoso y alteraciones cardíacas se ha descrito tanto en formas recesivas de MAVD (Naxos) como en formas recesivas de MCD26.
Estas proteínas desempeñan, por tanto, una función estructural semejante a otras descritas en la génesis de la MCD27.
Alteraciones en al acoplamiento excitación-contracción
El receptor cardíaco de la rianodina (RYR2) forma parte de la estructura que regula los canales del calcio del retículo sarcoplasmático. El funcionamiento correcto de estos canales es fundamental para el acoplamiento excitación-contracción y la homeostasis del calcio en los miocitos. Se han descrito 6 mutaciones del RYR2, en distintas familias; 3 codifican el aminoácido terminal y las otras 3, el centro de la proteína de unión28,29. En estas familias se describe una susceptibilidad mayor a las taquicardias ventriculares inducidas por el ejercicio.
En 1995 Rampazzo30 describe un segundo locus asociado a lo que él denomina una variante nueva de MAVD, la MAVD2. Ésta se caracteriza por presentar taquicardias ventriculares inducidas tras estímulo catecolaminérgico. Se han descrito mutaciones en este gen en la taquicardia ventricular polimórfica catecolaminérgica, la taquicardia ventricular polimórfica familiar y la MAVD229,31,32. Éstas, a diferencia de la MAVD2, no presentan sustitución fibroadiposa. El RYR2 alterado aumentaría la concentración citosólica de calcio, y desencadenaría la muerte celular programada15,33-38; asimismo, tendría lugar un desacoplamiento excitación-contracción que promovería arritmias.
La progresiva descripción de mutaciones causales de la MAVD abre el abanico tanto al entendimiento del amplio espectro de presentación de la enfermedad, y su variabilidad interfamiliar e intrafamiliar, así como a la controversia, ya que algunas entidades quizá no debieran contemplarse bajo la denominación sindrómica de MAVD. En particular, las taquicardias inducidas por el ejercicio se perfilan como una entidad independiente de la MAVD.
La unión de los esfuerzos para realizar un banco genético11,20 será indudablemente lo que marcará la progresión en el conocimiento y la caracterización de la MAVD.
PRESENTACIÓN CLÍNICA E HISTORIA NATURAL
La MAVD se manifiesta fundamentalmente en la adolescencia o en la edad adulta, y afecta más frecuentemente a los varones. La prevalencia varía ampliamente según las series descritas y existe controversia acerca de la distribución geográfica de la enfermedad. En regiones italianas del Véneto se estima una prevalencia de 1 caso por 1.000 o 10.000 personas. Corrado et al39,40 determinan que puede ser causa de hasta el 20% de las muertes súbitas (MS) en adultos jóvenes, y es la causa más frecuente de MS en atletas italianos. En EE.UU. representaría el 5% de las MS en menores de 65 años41 y sería causante del 3-4% de las MS en atletas42. Podría ser una causa frecuente de muertes perioperatorias en sujetos sin evidencia de cardiopatía estructural previa43-45. En nuestro país sería una causa frecuente de MS en varones jóvenes46.
Las manifestaciones clínicas son variables y en función de la inestabilidad cardíaca y de la disfunción ventricular progresiva. Las manifestaciones clínicas varían desde pacientes asintomáticos, MS como primera manifestación, arritmias ventriculares y supraventriculares hasta insuficiencia cardíaca derecha o biventricular.
En la MAVD se ha descrito la presencia de un desequilibrio en la inervación adrenérgica como posible coadyuvante en la génesis de las arritmias. De este modo aumentaría la propensión a arritmias ventriculares en situaciones de exposición a las catecolaminas, especialmente durante el ejercicio47.
Aunque la información en relación con la historia natural es limitada, en general se admiten 4 estadios.
1. La fase temprana o silente, generalmente asintomática, aunque el debut puede manifestarse con MS48.
2. Fase inestable con predominio de arritmias sintomáticas, generalmente con morfología de bloqueo completo de rama izquierda, altamente sugestivas de origen ventricular derecho.
3. Fase de fallo ventricular derecho con relativa conservación del ventrículo izquierdo (VI).
4. Fase final con progresiva dilatación biventricular, a menudo indistinguible de la MCD. Las complicaciones más frecuentes en este estadio son las tromboembólicas y la fibrilación auricular49.
Implicación del ventrículo izquierdo
La afección del VI conlleva, en la mayor parte de los casos, una progresión severa de la enfermedad. Series con seguimiento a largo plazo esclarecen y delimitan la participación del VI en la llamada MAVD. En una serie de 42 pacientes, se encontró afección ventricular izquierda en el 76% de ellos. En esta serie la afección de VI fue dependiente de la edad y se asoció con acontecimientos arrítmicos, cardiomegalia más severa, infiltrados inflamatorios e insuficiencia cardíaca50.
También se han descrito casos de «miocardiopatía arritmogénica del VI» con afección ventricular izquierda única de características anatomopatológicas idénticas a la del lado derecho51,52.
DIAGNÓSTICO
El diagnóstico definitivo de la MAVD requiere la confirmación anatomopatológica de la sustitución fibroadiposa transmural, mediante muestras quirúrgicas o necrópsicas. La naturaleza parcheada y progresiva de la enfermedad hace que la biopsia endomiocárdica tenga una utilidad diagnóstica limitada.
No existe una única prueba para establecer el diagnóstico de MAVD3,7. Éste se establece tras una evaluación funcional, morfológica y electrocardiográfica mediante la cual se determinan los criterios mayores y menores actualmente reconocidos (tabla 2).

Se deben cumplir 2 criterios mayores, 1 criterio mayor y 2 menores o bien 4 menores. Dichos criterios han probado su utilidad de manera prospectiva.
La tabla 3 expone los criterios propuestos por Hamid et al53 para aumentar la sensibilidad diagnóstica de los familiares de primer grado.

En la actualidad están en marcha varios registros internacionales longitudinales y prospectivos, el europeo49,50 y el norteamericano54, que entre otros objetivos pretenden analizar la validez de los criterios diagnósticos actuales y añadir las aportaciones de las asociaciones fenotipo-genotipo para mejorar la rentabilidad diagnóstica.
PRUEBAS DIAGNÓSTICAS
Electrocardiograma
La naturaleza progresiva de la enfermedad se ha demostrado en estudios prospectivos. Así, el electrocardiograma (ECG) inicial puede ser normal. En una serie de 74 pacientes con taquicardia ventricular (TV) y MAVD, el 40% de los pacientes presentaba un ECG normal en el primer año tras el episodio de TV. Posteriormente, sólo el 8% a los 5 años y ninguno más allá de los 6 años de seguimiento55.
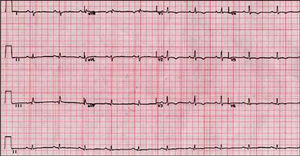
Las alteraciones del ECG más frecuentes son la inversión de la onda T (V1-V3), presente hasta en el 50% de los sujetos56. La afección más allá de V3 indica afección adicional del VI57 (fig. 1).

Fig. 1. Electrocardiograma de un paciente con miocardiopatía arritmogénica del ventrículo derecho y afección ventricular izquierda.
Existen diferentes anomalías de la despolarización descritas en la MAVD: el bloqueo de rama derecha incompleto es más frecuente (18%) que el completo (15%); la prolongación del QRS más de 110 ms en V1 y V2, un hallazgo más específico58, y la aparición de las ondas épsilon (fig. 2). Éstas se identifican en el 30%59, pero pueden pasar inadvertidas. La sensibilidad para su detección se puede aumentar con una adecuada preparación de la piel y obteniendo el ECG a doble velocidad y doble amplitud60. Se observan al final del QRS y al inicio del ST y corresponden a potenciales eléctricos retrasados de pequeña amplitud originados en las áreas de tejido sano rodeadas de infiltrado fibroadiposo61.

Fig. 2. Onda épsilon en la derivación V1.
La presencia de potenciales tardíos en el ECG promediado de señales (EPS) es equivalente a las ondas épsilon, y se relaciona con fibrosis miocárdica. Del 50 al 80% de los pacientes con TV presentan alteraciones en el EPS62,63, aunque esta prueba puede ser normal en casos con afección muy localizada.
Ecocardiografía
Es la primera técnica de imagen a utilizar al tratarse de un método no invasivo que permite analizar la progresión de la enfermedad, estudiar a los familiares y realizar un diagnóstico diferencial con otras entidades que cursen con arritmias y dilatación del VD. Entre éstas hay que incluir las comunicaciones interauriculares, las valvulopatías tricuspídea o pulmonar, el infarto de VD, la ausencia congénita de hemipericardio derecho, la anomalía de Ebstein y el retorno pulmonar anómalo parcial64,65.
Los hallazgos ecocardiográficos descritos incluyen la dilatación y la hipocinesia del VD, aneurismas telediastólicos y discinesia inferobasal. Debido a su carácter segmentario, la función debe evaluarse en diferentes zonas del VD. Es de utilidad reconocida la razón entre los tamaños ventriculares derecho e izquierdo. Otros hallazgos son un aumento de la ecogenicidad de la banda moderadora, trabéculas prominentes en el ápex66 y prolapso de la válvula tricúspide.
Con el avance de las técnicas de imagen y el desarrollo de software para el análisis objetivo, la ecocardiografía puede ayudar a detectar alteraciones tempranas así como de la progresión de la enfermedad. Las alteraciones del patrón de motilidad diastólica del anillo tricúspide obtenidas mediante Doppler tisular67 también se consideran de utilidad.
La beneficio de los contrastes ecocardiográficos para el diagnóstico de la MAVD está aún por demostrar, pero los resultados preliminares parecen prometedores.
Ventriculografía derecha
Esta técnica se considera el «patrón de oro» para la evaluación de la función del VD59,66. En las series descritas, la especificidad llega hasta el 90% para evidenciar áreas de acinesias en el llamado triángulo de la displasia. No obstante, no es un método difundido debido a la variación interobservador y al tratarse de una técnica invasiva.
Resonancia magnética cardíaca
La resonancia magnética cardíaca permite valorar anomalías tanto funcionales como estructurales. Es una técnica no invasiva de alta sensibilidad y no limitada por la ventana acústica. Además aporta la posibilidad de caracterización tisular. Idealmente ofrece inmejorables ventajas; puede caracterizar fibrosis, lo cual tiene potencialmente una utilidad diagnóstica en las formas iniciales, caracterizar la infiltración grasa y detectar las anomalías estructurales (fig. 3). La identificación de tejido adiposo en el miocardio del VD no implica de por sí el diagnóstico de MAVD6, ya que los individuos sanos presentan también infiltración grasa, fundamentalmente en la cara anterior del VD. Es una técnica ventajosa; sin embargo, presenta diferentes limitaciones, debidas a la variación interobservador61,68,69, a las propias del VD, en el que, al estar reducido el espesor parietal, la resolución espacial resulta a veces insuficiente para cuantificar el grosor ventricular (artefactos por el movimiento), y a la dificultad de evaluar a los pacientes con arritmias o simplemente numerosas extrasístoles.

Fig. 3. Imagen de resonancia magnética cardíaca. Abolladuras de la pared libre del ventrículo derecho. VD: ventrículo derecho; VI: ventrículo izquierdo.
Por tanto, es una técnica en proceso de evaluación cuyos resultados deben ser considerados, por el momento, en conjunto con otras pruebas diagnósticas.
Medicina nuclear
Al margen de las aportaciones obvias para evaluar los volúmenes y la función ventriculares, a escala molecular la gammagrafía isotópica con I-meta-yodo-bencilguanidina ha permitido identificar defectos en la inervación simpática70. Asimismo, otros estudios han demostrado la existencia de un menor número de receptores postsinápticos betaadrenérgicos en el miocardio47.
Biopsia endomiocárdica
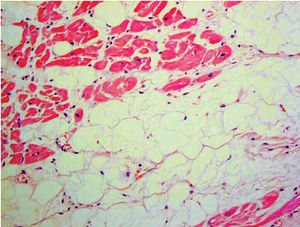
El diagnóstico histológico es definitivo71 (fig. 4). Sin embargo, la utilidad de la biopsia endomiocárdica es muy controvertida debido al carácter segmentario de la enfermedad, la escasa afección del septo (lugar habitual para la obtención de la muestra), una alta tasa de complicaciones (taponamiento y perforación) en relación con el adelgazamiento de la pared miocárdica y la mayor dificultad técnica (frecuentemente se obtienen muestras de localizaciones no habituales)7.

Fig. 4. Muestra anatomopatológica: infiltración fibroadiposa del ventrículo derecho.
Electrocardiograma promediado de señales, Holter y prueba de esfuerzo
Las áreas de conducción lenta generan pospotenciales tardíos. La prevalencia de estos potenciales varía según los estudios con una sensibilidad del 45% y alta especificidad (95%)72. La dispersión del QT > 40 ms, que mide la no homogeneidad de la repolarización ventricular, ha sido propuesta en combinación con el EPS para identificar los pacientes con MAVD con TV de origen derecho.
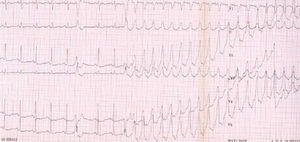
La prueba de esfuerzo y el registro Holter ponen de manifiesto arritmias, que constituyen criterios diagnósticos. Sin embargo, el valor pronóstico de un resultado normal es muy limitado (fig. 5).

Fig. 5. Taquicardia ventricular con morfología de bloqueo de rama izquierda desencadenada en la prueba de esfuerzo.
REGISTROS INTERNACIONALES
La MAVD está sujeta en la actualidad a intensa investigación. Están activos 2 registros principales: el norteamericano (www.arvd.org) y el europeo (www.arvd.net). Los objetivos y los protocolos de estudios pretenden ampliar al máximo la información disponible sobre la MAVD.
TRATAMIENTO, ESTRATIFICACIÓN DE RIESGO Y PRONÓSTICO
La MAVD es una causa común de MS. La aparición de MS no tiene relación con la progresión de la enfermedad, es decir, puede ser la primera manifestación. Aunque no existen todavía series prospectivas de supervivencia, la tasa de mortalidad anual sin tratamiento es del 2,5-3% y del 1% en individuos en tratamiento farmacológico73,74.
El tratamiento debe ser individualizado. Las distintas opciones terapéuticas incluyen el tratamiento fármacológico, el desfibrilador automático implantable (DAI), la ablación con radiofrecuencia y el trasplante.
No se dispone de un tratamiento farmacológico que haya demostrado, solo o en asociación con otros fármacos, ser totalmente efectivo en la protección frente a la MS. Para el tratamiento sintomático de las arritmias, fundamentalmente desencadenadas tras un estímulo catecolaminérgico, han demostrado especial eficacia los bloqueadores beta75,41.
El uso de fármacos tales como el sotalol y la amiodarona, sola o en asociaciones con bloqueadores beta, ha demostrado su efectividad en el tratamiento de las arritmias sintomáticas61,76. En particular, un estudio realizado con sotalol demostró que a dosis elevadas era capaz de suprimir las arritmias ventriculares hasta en el 84% de los pacientes que habían presentado arritmias ventriculares inducibles durante el estudio electrofisiológico77.
En los pacientes de riesgo elevado se debe considerar la colocación de un DAI.
Un reciente estudio prospectivo78 de 132 pacientes con MAVD a los que se les implantó un DAI incluyó las siguientes indicaciones: historia previa de parada cardíaca, TV con compromiso hemodinámico (síncope o colapso circulatorio), TV sin compromiso hemodinámico, TV no sostenida detectada en el Holter o en la prueba de esfuerzo o historia familiar de más de un familiar con MS. Tras un seguimiento medio de 3,3 meses, se registraron 4 muertes. Una arrítmica, tras fibrilación ventricular (FV) refractaria, 2 relacionadas con infección de la bolsa del DAI y otra tras insuficiencia cardíaca progresiva. Además, durante el seguimiento fue necesario realizar 3 trasplantes, 2 por fallo biventricular y 1 debido a FV intratable. De los 132 pacientes, 64 (48%) recibieron un total de 1.271 descargas apropiadas. La tasa de descarga en el grupo de estudio fue del 15% anual. El intervalo desde la implantación hasta la primera descarga fue muy variable, de 2 meses a 8 años. No hubo diferencias significativas en cuanto a la utilización de fármacos entre el grupo con descargas apropiadas y los 68 (52%) pacientes que no sufrieron descarga alguna.
El análisis estadístico señaló los siguientes posibles marcadores de riesgo para fibrilación/aleteo ventricular: parada cardíaca previa o TV con compromiso hemodinámico, edad temprana, y fracción de eyección deprimida del VI. El síncope de causa inexplicada casi alcanzó significación estadística. Los pacientes que recibieron un DAI por historia familiar de MS o por TV sin repercusión hemodinámica no presentaron resultados beneficiosos significativos. En el caso de la historia familiar, puede estar en relación con el pequeño número de pacientes que recibieron un DAI debido a esta única causa.
Los estudios retrospectivos han señalado distintos factores en relación con la MS: edad joven, historia de síncope de causa no explicada, historia de MS familiar, la práctica de deportes competitivos, la dilatación difusa ventricular derecha y la extensión al VI79-81 (tabla 4).
TABLA 4. Factores propuestos en relación con el incremento del riesgo de muerte súbitaa
Respecto a las dificultades técnicas, es importante recalcar que la zona de colocación del electrodo es crucial en la apropiada detección de arritmias ventriculares82.
Otras opciones terapéuticas invasivas incluyen la ablación por radiofrecuencia y el tratamiento quirúrgico de las arritmias.
La ablación por radiofrecuencia tiene su lugar en casos refractarios aunque, dados el grosor del VD y el carácter progresivo de la MAVD, tiene un uso limitado. En cuanto a su valor pronóstico, la no inducibilidad de TV con o sin terapia farmacológica no disminuyen el riesgo de MS83.
El tratamiento quirúrgico de las arritmias para los casos refractarios implica la realización de ventriculotomía o bien aislamiento del VD para aislar el circuito causante de la macrorreentrada2. Sin embargo, el riesgo de complicaciones, la elevada tasa de recurrencias de las arritmias ventriculares y las alteraciones hemodinámicas subsiguientes hacen que su uso sea cada vez más limitado. Sin embargo, el trasplante es una opción a tener en cuenta en las fases dilatadas de la enfermedad.
Ejercicio y muerte súbita
La práctica de deportes competitivos se ha relacionado con incremento de la MS84. Existe, sin embargo, una gran variabilidad en cuanto a la incidencia de MAVD en las series necrópsicas, que depende de la localización, así como de las circunstancias en que ésta sucede. Datos recientes85 de una serie retrospectiva de 200 necropsias de fallecidos súbitamente con MAVD señalan que la aparición más frecuente de la MS es durante actividades sedentarias. Sin embargo, la MAVD es una causa frecuente de MS, especialmente en atletas varones (≤ 35 años)39,42,46 de acuerdo con las repetidas observaciones de distintos grupos. El mismo grupo de Padua78 publica una de las pocas series prospectivas en la que investiga acerca de la aparición de MS en atletas jóvenes de ambos sexos en comparación con una población similar que no practica deportes competitivos. Los resultados indican que el deporte no es per se la causa de una mayor mortalidad, sino que actúa como un desencadenante para la parada cardíaca en las siguientes entidades: MAVD, enfermedad cardiovascular prematura y anomalías coronarias congénitas.
CONCLUSIÓN
La MAVD es una miocardiopatía cuya complejidad se ha visto ampliada a la luz de la progresión de los descubrimientos recientes. Aunque su incidencia se ha estimado en 1:5.000, su carácter familiar y su patrón hereditario hacen que su correcto manejo requiera un abordaje multidisciplinario y repetido de los familiares, lo que aumenta considerablemente el tamaño epidemiológico del problema. Los esfuerzos internacionales por aumentar la información disponible a nivel clínico, diagnóstico y genético requieren la cooperación de la mayor parte de los países. Por último, aunque el tratamiento farmacológico ha demostrado ser eficaz en la mayoría de los pacientes, el uso del DAI debe tenerse en cuenta especialmente entre aquellos con un perfil de riesgo elevado.
Financiación: José M. García-Pinilla disfrutó de la beca SEC para estancias cortas en el extranjero 2002. María T. Tomé Esteban disfrutó de la beca SEC para formación de posgrado en el extranjero 2002 y en la actualidad es Clinical Research Fellow financiada por la British Heart Foundation.
Correspondencia: Prof. W.J. McKenna.
Cardiology in The Young. The Heart Hospital.
16-18 Westmoreland Street. London W1G 8PH. United Kingdom.
Correo electrónico: william.mckenna@uclh.org