El término amiloidosis cardiaca hace referencia a la afección del corazón como consecuencia del depósito de amiloide en el tejido cardiaco, ya sea en el contexto de una afección sistémica o de una forma localizada. Diversas proteínas proamiloidóticas pueden dar lugar a depósitos amiloides en el corazón. Cada una de las amiloidosis producidas por estas proteínas presenta evolución, diagnóstico y tratamiento específicos, así como una clínica (cardiaca y extracardiaca) más característica. Dado que la primera manifestación de los pacientes con amiloidosis puede deberse a la afección cardiaca, el cardiólogo puede ser el primer profesional que atienda a estos pacientes y debe plantearse este diagnóstico siempre. En esta revisión presentamos, desde el punto de vista del cardiólogo y a la luz de nuestra experiencia, las características de las diferentes amiloidosis que pueden cursar con afección cardiaca revisando detalladamente cuándo y cómo establecer su diagnóstico. Además, repasamos el manejo terapéutico en estos pacientes tanto en lo referente a la afección cardiaca como a la enfermedad de base productora de la proteína amiloidótica.
Palabras clave
Amiloidosis es un término genérico que hace referencia al depósito extracelular de fibrillas anormales insolubles compuestas por diferentes subunidades de bajo peso molecular (entre 5 y 25 kDa). Estos depósitos proceden de proteínas solubles que, tras sufrir cambios conformacionales, adoptan una estructura predominante de hoja plegada beta alineadas de forma antiparalela1.
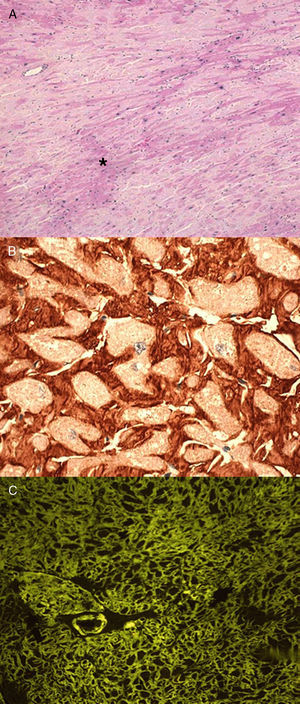
Anatomopatológicamente, los depósitos amiloides aparecen como material hialino que se tiñe con rojo Congo (dando refringencia verde bajo luz polarizada), tioflavina T (produciendo una intensa fluorescencia amarillo-verdosa) y azul Alcián (tinción verde) como se muestra en la Figura 12.

Figura 1. Biopsias cardiacas de paciente con amiloidosis cardiaca hereditaria por transtiretina. A: tinción de hematoxilina-eosina (×200); los depósitos de amiloide (*) aparecen como material amorfo entre los miocitos. B: inmunohistoquímica para detección de transtiretina (×400); depósitos color teja perimiocitarios. C: tinción de tioflavina T (×200); depósitos amarillo-verdosos situados alrededor de los miocitos que se corresponden con depósitos de sustancia amiloide. Modificado de García-Pavía et al 2 , cortesía de la Dra. C. Salas, del Hospital Universitario Puerta de Hierro, Madrid, España.
Se conocen más de 20 proteínas que pueden producir depósitos amiloides en distintos tejidos del organismo y dar lugar a diversas afecciones como la enfermedad de Alzheimer o las enfermedades por priones (Tabla 1).
Tabla 1. Proteínas amiloides humanas
| Amiloide | Precusor | S/L | Tipo clínico |
| AL | Inmunoglobulina ligera | S, L | Amiloidosis primaria |
| AH | Inmunoglobulina pesada | S, L | Amiloidosis primaria |
| ATTR | Transtiretina | S, L | Familiar senil |
| Aβ2M | Microglobulina β2 | S | Asociada a diálisis |
| AA | Amiloide A sérico | S | Asociada a infección e inflamación crónica |
| AApoAI | Apolipoproteína A-I | S | Familiar, nefropatíaDepósito senil en íntima aórtica |
| AApoAII | Apolipoproteína A-II | S | Familiar, nefropatía |
| AGel | Gelsolina | S | Familiar, neuropatía (síndrome de Meretoja) |
| ALys | Lisozima | S | Familiar, nefropatía |
| AFib | Fibrinógeno α | S | Familiar, nefropatía |
| ACys | Cistatina C | S | Familiar, hemorragia cerebral (tipo islandés) |
| ABri/ADan | Proteína precursora ABri (ABriPP) | S | Familiar, demencia (tipo británico y danés) |
| Aβ | Proteína precursora amiloide (APP) | L | Alzheimer |
| APrP | Prion (PRP) | L | Encefalopatía espongiforme |
| ACal | Procalcitonina | L | Tumor de células C tiroideas |
| AIAPP | Polipéptido islotes amiloides | L | Insulinomas, DM2 y edad |
| AANF | Péptido natriurético auricular (ANP) | L | Amiloidosis auricular asociada a la edad |
| APro | Prolactina | L | Prolactinomas, edad |
| AIns | Insulina | L | Depósitos locales asociados a bombas de insulina |
| AMed | Lactadherina | L | Depósito senil en media aórtica |
| AKer | Queratoepitelina | L | Familiar, distrofia corneal |
| ALac | Lactoferrina | L | Amiloidosis corneal |
| Semenogelina * | L | Depósitos seniles en vesículas seminales |
DM2: diabetes mellitus tipo 2; L: localizada; S: sistémica.
* Todavía no tiene nomenclatura asignada.
Según los órganos afectados, las amiloidosis se pueden clasificar en formas sistémicas o formas localizadas3. En las amiloidosis sistémicas, los depósitos se producen en múltiples órganos, en las paredes vasculares y en el tejido conectivo, y dan lugar a una clínica de afección multiorgánica. En las formas localizadas, los depósitos se encuentran circunscritos a un solo órgano o tejido y, por lo tanto, su clínica está limitada al sistema del que forme parte ese órgano o tejido.
El término amiloidosis cardiaca hace referencia a la afección cardiaca como consecuencia del depósito amiloideo en el tejido cardiaco, ya sea en el contexto de una afección sistémica (como es más frecuente) o de una forma localizada.
Sólo algunos de los precursores amiloidóticos producen afección cardiaca y su variada naturaleza hace que reconocerla y tratarla sea una tarea nada sencilla (Tabla 2).
Tabla 2. Subtipos de amiloidosis que afectan significativamente al corazón
| Tipo de amiloidosis | Proteína | Afección cardiaca | Mediana de supervivencia (meses) | Clínica extracardiaca habitual | Tratamiento |
| Primaria (AL) | Inmunoglobulina ligera | 50% | 13 (4 si IC al diagnóstico y sin tratamiento) | Nefropatía, proteinuria, disfunción autonómica, STC, neuropatía, macroglosia, púrpura | Quimioterapia+TMO |
| Secundaria (AA) | Amiloide A sérico | 5% | 24,5 | Nefropatía, proteinuria, hepatomegalia | Tratamiento de proceso inflamatorio/infeccioso subyacente |
| Hereditaria TTR (ATTR) | Transtiretina | Variable, en función de la mutación | 70 | Neuropatía, disfunción autonómica | Trasplante hepático |
| Hereditaria Apo-AI (AApoAI) | Apolipoproteína A-I | Variable, en función de la mutación | Sin datos | Nefropatía | Trasplante hepático |
| Hereditaria fibrinógeno A (AFib) | Fibrinógeno | Variable, rara | Sin datos | Nefropatía | Trasplante hepático |
| Senil (ATTR) | Transtiretina | 100% | 75 | STC | Soporte |
IC: insuficiencia cardiaca; STC: síndrome del túnel carpiano; TMO: trasplante de médula ósea.
Dado que la clínica cardiaca puede ser la primera manifestación de estas complejas e interesantes enfermedades, el cardiólogo puede ser el primer profesional que se enfrente a estos pacientes, y de su habilidad para identificar y enfocar el diagnóstico depende el inicio precoz del tratamiento.
Como enfermedad compleja que presenta patrón familiar en algunos casos y clínica cardiovascular y extracardiaca, en esta revisión repasaremos, desde el punto de vista del cardiólogo, las características clínicas de la amiloidosis cardiaca y la estrategia diagnóstica a emplear para reconocerla y tratarla en función del subtipo.
AMILOIDOSIS COMO ENFERMEDAD CARDIACAAunque varios tipos de amiloide pueden infiltrar el corazón, sólo la variedad senil, la secundaria (AA), la primaria (AL) y algunas formas de las hereditarias (ATTR, AApoA-I y AFib) pueden producir clínica cardiovascular significativa.
El patrón infiltrativo cardiaco es similar en todas ellas y puede afectar tanto a la función contráctil como al flujo vascular y a la conducción eléctrica.
Los depósitos se distribuyen en forma de agregados nodulares con ramificaciones que envuelven y aíslan a los miocitos. En las fases iniciales, los depósitos producen una disfunción diastólica leve, pero según progresan se produce un engrosamiento de las paredes, con empeoramiento de la relajación y la distensibilidad del ventrículo. El aumento de presiones origina una fisiología restrictiva en las fases más avanzadas y una dilatación importante de las aurículas4. Según progresa la enfermedad, se produce necrosis de los miocitos (en parte por efecto tóxico directo del amiloide)5 y desarrollo de fibrosis intersticial. Como resultado de todos estos fenómenos, en las fases avanzadas de la enfermedad, puede haber deterioro de la función sistólica.
A esto contribuye también la isquemia resultante de la infiltración amiloide de la microvasculatura6. La afección difusa de la microvasculatura genera numerosos focos endomiocárdicos de isquemia y microinfartos. Sorprendentemente, las arterias epicárdicas no suelen presentar afección significativa.
Los depósitos en el tejido de conducción no son habituales, aunque la fibrosis perivascular secundaria a la isquemia sí suele afectar al nodo sinusal y el haz de His7.
AMILOIDOSIS COMO ENFERMEDAD GENÉTICALa importancia de la herencia en la expresión de las enfermedades amiloidóticas se conoce desde hace años8. Algunas amiloidosis se deben en exclusiva a defectos genéticos pero, además, en el desarrollo de algunas amiloidosis adquiridas probablemente influyan otros factores también determinados genéticamente.
Tres tipos de anormalidades genéticas se han identificado en relación con las proteínas amiloidóticas: polimorfismos9, mutaciones10 y modificaciones postraslacionales determinadas genéticamente11.
Se han encontrado grandes diferencias en la presentación y el curso clínico de la amiloidosis entre sujetos con el mismo defecto genético12. Este hecho indica, por lo tanto, que los factores ambientales y los genes modificadores tienen un importante papel en la expresión de esta enfermedad.
La identificación de los pacientes cuya amiloidosis se debe a un defecto genético tiene gran importancia, ya que modifica el tratamiento y además tiene gran trascendencia para los familiares2. Dado que la presencia de bandas monoclonales (gammapatía monoclonal de significado incierto) no es infrecuente en la población general, siempre hay que tener en cuenta las causas hereditarias de amiloidosis y excluirlas adecuadamente. En un centro inglés de referencia, hasta casi el 10% de los pacientes sin antecedentes familiares compatibles con amiloidosis hereditaria y con diagnóstico de amiloidosis AL presentaron mutaciones en alguno de los genes relacionados con amiloidosis hereditarias (principalmente fibrinógeno y transtiretina [TTR])13.
TIPOS DE AMILOIDOSIS QUE PRODUCEN AFECCIÓN CARDIACATal como hemos comentado anteriormente, sólo algunas proteínas amiloidóticas producen afección cardiaca (Tabla 2). Sus variables presentación y curso clínico hacen de ellas entidades independientes14. El cardiólogo debe conocerlas todas, pues cuando se enfrente a un paciente con amiloidosis, el diagnóstico diferencial quedará restringido a una de ellas.
Amiloidosis ALEs la forma más común en países desarrollados y se produce por depósito del dominio variable, o parte de él, procedente de una forma monoclonal de una cadena de inmunoglobulinas ligeras. Prácticamente cualquier discrasia que afecte a los linfocitos B (mieloma, linfoma, macroglobulinemia, etc.) puede producir esta proteína monoclonal y dar lugar a amiloidosis.
Aparece normalmente por encima de los 50 años, pero puede aparecer antes. La distribución por sexos es similar o con ligero predominio en los varones, según las series15, 16. La afección multiorgánica es lo habitual, aunque en el 5% de los casos el corazón es el único órgano clínicamente afectado15.
En el 90% de los casos hay depósitos cardiacos, pero sólo el 50% de los pacientes presentan síntomas o signos de afección cardiaca en el momento del diagnóstico.
La afección cardiaca marca el pronóstico, con una mediana de supervivencia total de 13 meses, que baja a 4 meses sin tratamiento si ya hay signos de insuficiencia cardiaca (IC) en el momento del diagnóstico17. Incluso si la afección de otro órgano o sistema predomina, la afección cardiaca es el peor factor pronóstico17.
Dado que el pronóstico de la amiloidosis AL es sensiblemente peor que en otros tipos de amiloidosis cardiacas14, se ha señalado que las cadenas ligeras circulantes pudieran tener un efecto tóxico directo5.
Amiloidosis AASe produce por depósito de proteína amiloide A sérica (AAs), un reactante de fase aguda que se encuentra elevado persistentemente en procesos infecciosos e inflamatorios crónicos.
En nuestro medio suele aparecer asociada a la artritis reumatoide, la fiebre mediterránea familiar, infecciones crónicas y la enfermedad inflamatoria intestinal.
La afección renal domina el cuadro clínico, con proteinuria e insuficiencia renal casi siempre. La supresión de la producción de AAs puede dar lugar a la disminución de los depósitos de amiloide y mejoría de la función renal18. La afección cardiaca es rara (5%) y, si la hay, suele ser leve19. La mediana de supervivencia es algo superior a los 24 meses4.
Amiloidosis hereditariasSe trata de condiciones autosómicas dominantes en las que los agregados amiloides son consecuencia del depósito de proteínas mutadas. De las distintas amiloidosis hereditarias sólo la apolipoproteína A-I, el fibrinógeno A y, con frecuencia mucho mayor, la TTR pueden depositarse en el corazón.
Apolipoproteína A-IEl gen de la apolipoproteína A-I (APOA1) se localiza en el cromosoma 1, y hasta la fecha se han descrito 16 mutaciones en él20. Las manifestaciones clínicas varían en función del tipo y la localización de la mutación en el gen20. Además de la afección cardiaca, las manifestaciones descritas incluyen: nefropatía, neuropatía, depósitos cutáneos, afección hepática y disfunción laríngea.
Fibrinógeno AEl gen (FGA) se localiza en el cromosoma 4 y, por el momento, todas las mutaciones amiloidóticas descritas (una decena) se encuentran en el exón 5. Todos los pacientes presentan, casi exclusivamente, afección renal. Es raro que la afección cardiaca sea significativa, aunque puede ser grave21. La expresividad de la enfermedad es muy variable, por lo que puede no haber antecedentes familiares, lo que dificulta el diagnóstico.
TranstiretinaLa TTR o prealbúmina, al igual que la apolipoproteína A-I y el fibrinógeno A, es una proteína sintetizada fundamentalmente en el hígado. Su gen (TTR) se localiza en el cromosoma 18, y en él se han descrito más de 100 mutaciones. Las distintas mutaciones dan lugar a fenotipos diferentes22. Se han comunicado formas neuropáticas, cardiacas, renales y oculares (por depósitos intravítreos). La afección cardiaca suele aparecer a partir de la quinta década y parece más frecuente en varones, lo que ha llevado a plantear el posible papel protector del sexo femenino contra el desarrollo de esta enfermedad16. Una de las mutaciones más comunes es la Val30Met. Esta mutación es endémica en zonas de Portugal, Suecia y Japón, y su prevalencia en estas áreas puede alcanzar 1:600. Aunque la clínica predominante es neuropática, el corazón también puede estar afectado22. Otras mutaciones, como la Thr59Lys o Glu89Lys, dan lugar a afección predominantemente cardiaca con cuadros restrictivos graves22. Entre el 3 y el 4% de los individuos de raza negra en Estados Unidos son portadores de la mutación Val122Ile, que se asocia al desarrollo de amiloidosis cardiaca en mayores de 60 años23. En un amplio estudio de autopsias a pacientes con amiloidosis senil, el 23% de los individuos de raza negra eran portadores de esta mutación, mientras que no estaba presente en individuos de raza blanca23. Aunque la prevalencia de la amiloidosis causada por esta mutación es desconocida, probablemente sea una entidad infradiagnosticada por atribuir la hipertrofia cardiaca de estos pacientes a cardiopatía hipertensiva.
Amiloidosis senilEn las autopsias de pacientes ancianos, con frecuencia se encuentran depósitos amiloides tanto en las aurículas (formados por acumulaciones de péptido natriurético auricular) como en los ventrículos (donde, en cambio, proceden de la forma no mutada de la TTR). En la gran mayoría de los casos, estos depósitos no tienen ninguna significación clínica. Sin embargo, en algunos sujetos, los depósitos ventriculares son masivos y dan lugar al cuadro que conocemos como amiloidosis senil y se caracteriza por cardiomegalia e IC.
Es excepcional por debajo de los 60 años y puede alcanzar una prevalencia de hasta un 25-36% por encima de los 80 años24. Afecta casi exclusivamente a varones14 y, a diferencia de otras formas de amiloidosis, la afección de otros órganos es excepcional (exceptuando la presencia de síndrome del túnel carpiano).
Pese a la avanzada edad de los pacientes y la gran infiltración cardiaca, la IC es de más fácil control y su mediana de supervivencia es de 75 meses, muy superior a la de otros tipos de amiloidosis14. En cualquier caso, la muerte de estos pacientes suele estar en relación con la progresión de la IC y la aparición de arritmias.
CLÍNICA CARDIOVASCULARInsuficiencia cardiacaAunque los síntomas extracardiacos pueden ser muy variados, la clínica cardiaca está dominada por el desarrollo de IC diastólica con predominancia de signos congestivos. Aunque las presiones en el ventrículo izquierdo están elevadas, el edema de pulmón es infrecuente.
AnginaAlgunos pacientes sufren angina en relación con la infiltración amiloide vascular. En ocasiones la angina se acompaña también de claudicación mandibular25. Como se ha comentado previamente, los vasos afectados son normalmente intramiocárdicos y las arterias epicárdicas no suelen mostrar lesiones.
Síncope o presíncopeLa presencia de síncope o presíncope es habitual en la amiloidosis cardiaca y es el resultado de la combinación de la disfunción autonómica (que estos pacientes suelen presentar) y/o arritmias con un corazón con poca reserva funcional. Los depósitos alteran la regulación adrenérgica del corazón, así como la regulación basal y la respuesta cardiaca a la estimulación neurohormonal26. Frecuentemente se observa desaparición de la hipertensión arterial (HTA) si el paciente era previamente hipertenso, con desarrollo de hipotensión postural secundaria a la excesiva diuresis o la neuropatía autonómica15.
La aparición de síncope de esfuerzo tiene un pronóstico infausto, ya que es un marcador de miocardiopatía restrictiva severa y se asocia a una mortalidad elevada en los 3 meses siguientes, normalmente en relación con muerte súbita27.
ArritmiasLa presencia de fibrilación auricular es frecuente debido a la dilatación progresiva de las aurículas y la fisiología restrictiva28. Cuando la fibrilación auricular está presente se asocia a una alta incidencia de tromboembolias.
Aunque las arritmias ventriculares son frecuentes, son causa infrecuente de síncope y no suelen ser el síntoma de presentación29.
Pese a lo que se piensa habitualmente, los bloqueos auriculoventriculares de alto grado y la disfunción sintomática del nodo son raros30. Aunque el nodo sinusal puede estar afectado por los depósitos, los trastornos de la conducción son más propios del sistema His-Purkinje, con la función del nodo auriculovenricular normalmente preservada.
La muerte súbita, aunque frecuente en estos pacientes29, suele deberse más a disociación electromecánica que al desarrollo de arritmias ventriculares.
OtrosFormas más raras de presentación son el taponamiento cardiaco por depósito de amiloide en el pericardio31 o la acumulación desproporcionada de amiloide en el septo interventricular que asemeja una miocardiopatía hipertrófica32. Aunque la obstrucción dinámica del tracto de salida del ventrículo izquierdo puede ocurrir33, es rara. Se ha señalado que la distribución septal asimétrica es más propia de pacientes con formas hereditarias que con amiloidosis AL32.
Por último, destacar que un accidente cerebrovascular puede ser la primera manifestación en un número significativo de casos34. En la mayoría de las ocasiones, el origen del accidente cerebrovascular es cardioembólico en relación con la formación de trombos intracardiacos.
CLÍNICA EXTRACARDIACAAunque su presencia varía en función del tipo de amiloidosis, en nuestra experiencia, casi siempre hay algún otro órgano afectado, en mayor o menor grado. La presencia de síntomas extracardiacos es un elemento importante a la hora de plantear el diagnóstico de esta entidad y, por lo tanto, se debe investigar específicamente.
Múltiples signos dermatológicos se han relacionado con la amiloidosis cardiaca; destacan los depósitos cutáneos de amiloide, la casi patognomónica púrpura periorbitaria y la fragilidad capilar (que, junto con el déficit de factores de coagulación, da lugar a hematomas cutáneos)35.
La presencia de macroglosia (un 10-20% de los pacientes con amiloidosis AL), puede producir disgeusia o disfonía. La combinación de macroglosia y púrpura periorbitaria tiene baja sensibilidad (10-20%), pero alta especificidad para establecer el diagnóstico36.
En ocasiones los pacientes refieren la presencia de uñas quebradizas y de crecimiento lento, que orienta también hacia la naturaleza sistémica de la enfermedad.
Los síntomas neurológicos incluyen historia (incluso familiar) de síndrome del túnel carpiano, polineuropatía sensitiva y autonómica.
El malestar en el hipocondrio derecho puede deberse tanto a congestión hepática como a infiltración hepática por amiloide37.
Finalmente, la afección renal es la norma en determinadas amiloidosis (AA, AFib) y no infrecuente en otras (AL). La proteinuria, incluso en rango nefrótico (≥ 3 g/24 h), no es inhabitual y su comprobación es un test sencillo que puede levantar la primera sospecha del diagnóstico.
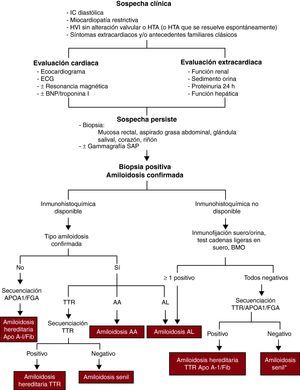
DIAGNÓSTICOSospecha clínicaEl cardiólogo debe plantearse el diagnóstico (Figura 2) de amiloidosis cardiaca ante todo paciente que presente IC diastólica, miocardiopatía restrictiva o engrosamiento de las paredes ventriculares en ausencia de alteración valvular o HTA (o HTA que se resuelve espontáneamente).

Figura 2. Proceso diagnóstico de amiloidosis cardiaca. APOA1: apolipoproteína A-I; BMO: biopsia de médula ósea; BNP: péptido natriurético cerebral; ECG: electrocardiograma; FGA: fibrinógeno A; HTA: hipertensión arterial; HVI: hipertrofia ventricular izquierda; IC: insuficiencia cardiaca; SAP: componente P de amiloide sérico; TTR: transtiretina. *Si se han descartado procesos inflamatorios/infecciosos crónicos que pudieran producir amiloidosis A.
La presencia de síntomas extracardiacos y/o antecedentes familiares clásicos (neuropatía, síndrome de túnel carpiano, IC cerca de la quinta década, etc.) también deben orientar al diagnóstico.
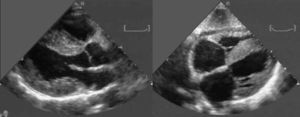
Evaluación cardiacaEcocardiogramaMediante ecocardiografía se puede encontrar diferentes signos de esta enfermedad, aunque los clásicos (Figura 3) sólo son propios de las fases avanzadas de la enfermedad38. El hallazgo más precoz es el engrosamiento de las pared ventricular izquierda (particularmente en ausencia de HTA), con evidencia de disfunción diastólica asociada38, 39. La presencia de paredes engrosadas, obviamente, tiene poca especificidad, ya que se encuentra también en otras afecciones cardiacas como la cardiopatía hipertensiva, la miocardiopatía hipertrófica y otras enfermedades infiltrativas del corazón (hemocromatosis, sarcoidosis, enfermedad de Fabry, etc.). La presencia de un patrón «granular» del miocardio se ha propuesto como un signo propio de esta entidad39, 40; sin embargo, su utilidad es limitada, ya que también aparece en otras causas de hipertrofia40, su sensibilidad es baja40 y sólo es valorable en ausencia de segundo harmónico. La disfunción diastólica, en cambio, es el hallazgo ecocardiográfico por antonomasia de esta enfermedad y está presente, en algún grado, en prácticamente todos los pacientes38. Los depósitos amiloides afectan a la distensibilidad del ventrículo, lo que se traduce en una disminución de la velocidad del flujo a través de la válvula mitral en la fase inicial de la diástole (disminución de la onda E) y un aumento en la fase tardía (aumento de la onda A) por mayor dependencia en la contracción auricular en el llenado ventricular. La disminución en la relación E:A es un signo precoz de la afección. Sin embargo, según los ventrículos se vuelven menos distensibles, la presión intrauricular aumenta y del mismo modo la velocidad inicial de llenado, dando lugar a una seudonormalización de la relación E:A. Finalmente, en fases avanzadas, lo habitual es encontrar un patrón de llenado restrictivo, con un tiempo de deceleración de la onda E reducido y baja velocidad de la onda A, unido a anormalidades en el flujo de las venas pulmonares41.

Figura 3. Ecocardiograma característico de paciente con amiloidosis por transtiretina. Izquierda: eje paraesternal longitudinal. Derecha: plano subcostal. Obsérvese el grosor aumentado del septo interventricular, ambos ventrículos y el septo interauricular. También es evidente un derrame pericárdico ligero. Cortesía del Laboratorio de Imagen Cardiaca del Policlinico S. Orsola-Malpighi, Bolonia, Italia.
El uso de Doppler tisular y strain rate puede ser de especial interés en esta enfermedad. Así, mediante Doppler tisular se encuentra reducción en las velocidades diastólicas tanto en fases tempranas como tardías de la enfermedad, y permite su identificación incluso cuando prácticamente no hay hipertrofia ventricular42. El strain rate permite documentar precozmente la afección cardiaca al detectar un deterioro en la función contráctil longitudinal43, y sus hallazgos se han relacionado incluso con el desarrollo posterior de IC y con la posibilidad de diferenciar distintos tipos de amiloidosis44.
Otros hallazgos ecocardiográficos incluyen la presencia de valvas engrosadas, derrame pericárdico ligero, dilatación biauricular y engrosamiento del septo interauricular. Aunque todos estos signos se describen en un 40-60% de los pacientes (con excepción del engrosamiento del septo interauricular, que es menos frecuente), se trata de series muy seleccionadas con pacientes que se encuentran en fases avanzadas de la enfermedad. La disfunción sistólica, por su parte, tampoco aparece hasta fases avanzadas.
ElectrocardiogramaLa serie más amplia descrita hasta la fecha de ECG en pacientes con amiloidosis confirmada mediante biopsia28 encontró que sólo el 46% de los pacientes presentaban el hallazgo clásico de bajos voltajes (amplitud de QRS ≤ 0,5 mV en todas las derivaciones de miembros o ≤ 1 mV en todas las precordiales). De hecho, se encontraron signos electrocardiográficos de hipertrofia ventricular (definida mediante criterios de Cornell o Sokolov) en el 16% de los pacientes. Otros hallazgos incluyeron la presencia habitual de patrón de seudoinfarto (sin infarto en el ecocardiograma) en el 47% de los pacientes. El 76% de los pacientes no presentaron ningún trastorno de la conducción, y sólo el 3% presentó bloqueos auriculoventriculares de segundo o tercer grado (el 21%, de primer grado) y el 14,5%, bloqueo de rama (el 9% de rama derecha y el 5,5% de rama izquierda). Fibrilación auricular o flutter sólo se encontró en el 10% de los pacientes. Ninguna de las variables electrocardiográficas tuvo implicaciones pronósticas en ese trabajo. Se ha evaluado la posibilidad de combinar los hallazgos del ECG y el ecocardiograma, y se halló que la combinación de bajos voltajes y engrosamiento ventricular era más sensible en el diagnóstico de amiloidosis que ambas pruebas por separado39, 45. En nuestra experiencia, la prevalencia de bajos voltajes es significativamente menor en pacientes con amiloidosis por TTR frente a pacientes con amiloidosis AL (el 30 frente al 50%). Esta observación subraya la importancia de ahondar en el diagnóstico ante cualquier sospecha clínica de amiloidosis, incluso en ausencia de los hallazgos «típicos».
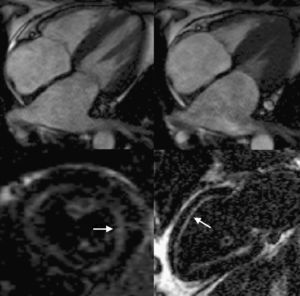
Resonancia magnéticaLa resonancia magnética (RM) permite una caracterización morfológica excelente (especialmente útil cuando hay limitaciones técnicas en las imágenes ecocardiográficas46), con alta reproducibilidad y la ventaja añadida de permitir el estudio mediante la técnica de realce tardío con gadolinio (Figura 4). El depósito de amiloide afecta específicamente a la cinética de distribución del gadolinio entre la sangre y el miocardio. Así, tras la administración de gadolinio se encuentra un mayor acortamiento del T1 subendocárdico y de la diferencia entre la señal en T1 de subendocardio y de sangre. Esta menor diferencia entre el subendocardio y la sangre se considera que refleja la rápida captación de gadolinio por los depósitos amiloides del miocardio y su desaparición rápida de la sangre. El patrón de captación de gadolinio en los pacientes con amiloidosis cardiaca se describió inicialmente como subendocárdico y general (sin afección circunscrita a un territorio coronario, como ocurre en la cardiopatía isquémica)47. Posteriormente se ha comprobado que el patrón de captación de gadolinio también puede ser parcheado localizado o transmural48, 49 y que incluso existen pacientes que, pese a presentar depósitos cardiacos de amiloide, no muestran captación alguna de gadolinio48, 49, 50. Cabe destacar, además, que la selección del tiempo de inversión T1 puede ser especialmente difícil en algunos casos y que, pese a emplear múltiples secuencias T1, no se logre anular apropiadamente la señal miocárdica, lo que impide determinar el patrón de captación de gadolinio del miocardio49. Algunos trabajos han relacionado la existencia de un patrón de captación general y subendocárdico con fases más avanzadas de la enfermedad49. De hecho, aunque no está establecido por completo el papel diagnóstico de la RM, la coexistencia de este patrón de captación «típico» junto con la demostración de amiloide en otro órgano puede ya interpretarse como sinónimo de afección cardiaca amiloidea y, por lo tanto, se puede evitar la biopsia cardiaca51, 52. Desgraciadamente, todavía desconocemos qué papel puede tener la RM en el diagnóstico en fases precoces de la enfermedad49, 51. En cuanto al papel pronóstico de la RM, no parece que la presencia y la extensión de los depósitos de gadolinio tengan influencia en la evolución de estos pacientes50, 53, 54, pero son necesarios trabajos más amplios, ya que se ha señalado que otras técnicas, como la cartografía T1, podrían caracterizar mejor la cantidad de amiloide intersticial y ofrecer importante información pronóstica54.

Figura 4. Resonancia magnética de 2 pacientes con amiloidosis. Imágenes superiores: planos de cine de cuatro cámaras en diástole (izquierda) y sístole (derecha). Obsérvese la importante hipertrofia ventricular con pobre contracción longitudinal y la gran dilatación biauricular. Imágenes inferiores: tras administración de gadolinio eje corto (izquierda) y apical de dos cámaras (derecha), que muestran captación de contraste a nivel subendocárdico (flechas). Cortesía del Dr. J. Moon, The Heart Hospital, Londres, Reino Unido.
Confirmación del diagnóstico de amiloidosis cardiacaLa confirmación del diagnóstico de amiloidosis cardiaca requiere la demostración de depósito amiloide en una biopsia, si bien esta no tiene que ser necesariamente cardiaca.
Si hay signos ecocardiográficos típicos y se demuestra el depósito de amiloide en otros tejidos, el diagnóstico de amiloidosis cardiaca puede darse como válido.
Otros tejidos, más accesibles que el cardiaco y habitualmente utilizados son la mucosa rectal55 (sensibilidad del 75-85%) o el aspirado de grasa abdominal, que es incluso más sensible (84-88%) y no tiene riesgo de sangrado o perforación56.
Nosotros, al igual que otros centros57, hemos utilizado en ocasiones la biopsia de glándula salival, con excelentes resultados incluso en pacientes con aspirado de grasa abdominal negativo.
Si la biopsia de otros tejidos es negativa y la sospecha persiste, la biopsia cardiaca es necesaria. Cuatro muestras endomiocárdicas aseguran una sensibilidad del 100% para la detección de la enfermedad58.
Diagnóstico del tipo de amiloidosisDado que el pronóstico y, especialmente, el tratamiento de esta enfermedad dependen del tipo de amiloidosis, una vez establecido el diagnóstico de amiloidosis cardiaca, es fundamental conocer frente a qué subtipo nos encontramos.
Existen diferentes técnicas inmunohistoquímicas59 y de inmunofluorescencia60 que permiten distinguir el tipo de material amiloide, si bien no están disponibles en todos los centros y, cuando lo están, no suelen incluir subtipos inhabituales de amiloidosis hereditarias. Tampoco distinguirán entre los dos tipos por depósito de TTR (amiloidosis senil y hereditaria por TTR), para lo que habrá que recurrir al análisis genético a fin de descartar alguna mutación.
Si no se ha podido realizar determinación de subtipo en la biopsia, habrá que descartar amiloidosis AL mediante la exclusión de una discrasia productora de cadenas ligeras. Para ello se recomienda realizar inmunofijación en suero y orina, más que electroforesis, ya que la cantidad de paraproteína puede ser pequeña y la inmunofijación tiene mayor sensibilidad. Los kits para detección de cadenas ligeras en plasma tienen todavía más sensibilidad (10 veces superior que la inmunofijación)61. Estos kits son un test cuantitativo que mide las concentraciones de cadenas ligeras libres kappa y lambda (normal, 3,3-19,4 y 5,7-26,3mg/dl, respectivamente) y la relación kappa/lambda (normal, 0,26-1,65)19. Como las cadenas ligeras se eliminan por vía renal, ambas estarán elevadas si hay afección renal, pero será el valor de la relación kappa/lamba lo que nos indique si hay producción monoclonal de una de ellas. Una relación kappa/lambda < 0,26 indica la existencia de una población clonal productora de cadenas lambda (lo más frecuente) y una relación > 1,65 indica una población productora kappa. Se ha demostrado que la combinación de una relación kappa/lambda anormal y una reacción de inmunofijación positiva tiene un 99% de sensibilidad para el diagnóstico de amiloidosis AL62. En estos casos, la biopsia de médula ósea nos dará la respuesta final acerca de la discrasia sanguínea productora de la paraproteína.
Es importante tener en cuenta que hasta en el 10% de los sujetos mayores de 70 años puede encontrarse de forma incidental una banda monoclonal en la inmunofijación («gammapatía monoclonal de significado incierto»). El kit para detección de cadenas ligeras en plasma dará normal en estos casos63 y habrá que realizar otras pruebas para excluir las otras formas de amiloidosis (incluyendo el análisis de ADN para diferenciar las hereditarias de la amiloidosis senil).
Un último comentario merecen las pruebas nucleares. Tradicionalmente la gammagrafía de detección del componente P de amiloide sérico (SAP) se ha empleado en centros especializados para evaluar la extensión sistémica de los depósitos de amiloide e incluso para monitorizar la respuesta al tratamiento64. Dado que el marcado del SAP no está disponible comercialmente, que esta técnica tiene poca sensibilidad en los pacientes con amiloidosis TTR y que, además, no permite caracterizar los depósitos en estructuras dinámicas como el corazón, su utilidad en la amiloidosis cardiaca es limitada64.
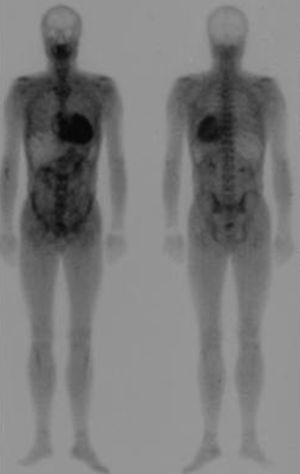
La gammagrafía con tecnecio difosfato (99mTc-DPD) se ha propuesto como una técnica interesante para distinguir entre los depósitos cardiacos de amiloide TTR frente a los de la amiloidosis AL65. Los corazones de los pacientes con amiloidosis TTR (ya sea hereditaria o senil) captarían 99mTc-DPD, mientras que los de aquellos con amiloidosis AL no lo captarían (Figura 5). Tras varios años aplicando esta técnica, hemos comprobado que, cuando la captación es abundante o nula, efectivamente nos permite discriminar las dos etiologías, pero que hasta un tercio de los sujetos con amiloidosis AL presentan captación ligera a nivel cardiaco66.

Figura 5. Gammagrafía con 99mTc-DPD en un paciente con amiloidosis por transtiretina. Se observa intensa captación del radiotrazador a nivel cardiaco con captación ósea atenuada.
TRATAMIENTOEl tratamiento de la amiloidosis cardiaca consta de dos facetas: por un lado, el tratamiento de los síntomas cardiacos y, por otro, el de la enfermedad de base productora de la proteína amiloidótica.
Tratamiento cardiacoLa base del tratamiento es la administración de diuréticos para controlar los síntomas congestivos. Si el paciente tiene afección renal importante, con disminución de la concentración de albúmina a consecuencia del síndrome nefrótico, pueden ser necesarias dosis muy altas de diuréticos. El control del peso y el balance hídrico diario (con ajuste de los diuréticos según las variaciones) es muy importante en estos pacientes para prevenir ingresos.
No existen datos acerca del uso de fármacos bloqueadores beta, pero asociados a la neuropatía autonómica que pueden tener estos pacientes, pueden causar hipotensión y bradicardia.
Los inhibidores de la enzima de conversión de angiotensina y los antagonistas del receptor de la angiotensina II suelen tolerarse pobremente (salvo en la amiloidosis senil) y deben usarse con cuidado, ya que incluso en dosis bajas pueden causar hipotensión severa (también en relación con la neuropatía autonómica).
Tanto la digoxina como algunos antagonistas del calcio se unen a los depósitos de amiloide y, por lo tanto, su administración es complicada, pues es muy difícil controlar sus concentraciones. Como resultado de la unión de la digoxina a los depósitos de amiloide, puede producirse intoxicación digitálica incluso con concentraciones séricas normales.
Debe iniciarse terapia anticoagulante si se documentan trombos intracardiacos, fibrilación auricular o ausencia de contracción auricular en el ecocardiograma67. Algunos autores recomiendan anticoagular sistemáticamente si la velocidad de la onda A transmitral es ≤ 20cm/s19.
La amiodarona suele tolerarse bien si se decide su uso para intentar mantener el ritmo sinusal.
Aunque la muerte súbita no es infrecuente, parece que se relaciona más con disociación electromecánica que con arritmias ventriculares, de modo que la utilidad del desfibrilador en esta población no está clara68. En la serie más larga publicada (19 receptores de desfibrilador)68 sólo 2 sujetos recibieron descargas apropiadas, mientras que ocurrieron 6 casos de muerte súbita por disociación electromecánica (uno en un paciente que sufrió previamente una descarga apropiada).
La indicación de marcapasos, por su parte, debe seguir las recomendaciones generales. Sin embargo, dado que en estos pacientes coexisten otros factores que agravan episodios de bajo gasto cardiaco (neuropatía autonómica e hipoalbuminemia), el umbral para implantar estos dispositivos suele ser muy bajo.
Tratamiento de la enfermedad de baseAmiloidosis ALEl tratamiento definitivo supone la administración de quimioterapia destinada a eliminar o controlar la discrasia productora de la paraproteína amiloidótica69, 70, 71. La quimioterapia puede detener la formación de amiloide y conducir a la regresión de los depósitos en muchos pacientes69, 70. Existen diversos regímenes utilizados en los pacientes con amiloidosis AL, entre ellos pautas que implican la utilización de nuevos fármacos como el rituximab o el bortezomib. La terapia intensiva con altas dosis de melfalán asociada al trasplante de médula ósea (TMO) es el procedimiento de elección en la amiloidosis AL, aunque acarrea una mortalidad alta (10-25%)19, 70. La afección cardiaca es uno de los mayores condicionantes de la respuesta satisfactoria a este tratamiento71, de modo que se considera que los pacientes con IC descompensada, fracción de eyección < 40% o presión arterial sistólica < 90mmHg no deben someterse a TMO. Además, los pacientes con afección significativa de dos o más órganos tienen una morbimortalidad elevada y, por lo tanto, no parecen buenos candidatos para este procedimiento72. En nuestra opinión, dada la complejidad de estos pacientes, para lograr un manejo óptimo es fundamental su tratamiento en centros con experiencia donde haya colaboración estrecha entre diferentes especialistas73.
Amiloidosis hereditariasEn estas enfermedades, el único procedimiento efectivo para tratar la fuente productora de la proteína amiloidótica es el trasplante hepático (TxH). Desde que en 1991 se hiciese el primer TxH a un paciente con amiloidosis hereditaria TTR, se han realizado más de 700 de estos trasplantes. El resultado suele ser bueno y el hígado de estos pacientes se puede emplear para TxH a receptores subóptimos (trasplante dominó). Desgraciadamente, se ha comprobado que el trasplante dominó puede conllevar el desarrollo de amiloidosis en el receptor74. En familias con varios miembros que han sufrido amiloidosis cardiaca y con otros miembros que son portadores de la mutación, hemos encontrado problemas en establecer cuándo es el momento óptimo para realizar el TxH que prevenga la afección cardiaca irreversible2. La opinión de los centros con más experiencia es que no debe realizarse el TxH hasta que no haya afección clínica significativa de un órgano o sistema.
Amiloidosis AAEl tratamiento de la enfermedad inflamatoria/infecciosa de base en la amiloidosis AA reduce las concentraciones plasmáticas de AAs y mejora el pronóstico espectacularmente18. Los nuevos fármacos biológicos (inhibidores del factor de necrosis tumoral y la interleucina 1) suprimen la respuesta aguda en pacientes con enfermedades autoinmunitarias y previenen el desarrollo de amiloidosis AA75. Por su parte, el tratamiento con colchicina en los pacientes con fiebre mediterránea familiar también previene el desarrollo de la enfermedad.
Trasplante cardiacoCuando la afección cardiaca es muy grave, el trasplante cardiaco (TxC) y los dispositivos de asistencia ventricular se convierten en la única alternativa para prolongar de forma significativa la supervivencia de estos pacientes. Sin embargo, los dispositivos de asistencia ventricular se han usado sólo de manera anecdótica en esta situación clínica, y el TxC se ha realizado esporádicamente ante la posibilidad de progresión de la enfermedad en otros órganos y recurrencia de la amiloidosis en el injerto.
Aunque muchos de los centros trasplantadores del mundo rechazan a estos pacientes, se trata de una opción factible si se comprueba que la afección amiloidótica significativa sólo afecta al corazón.
En pacientes con amiloidosis AL y afección aislada del corazón (por la que no podrían someterse a TMO), se ha realizado con éxito TxC seguido de altas dosis de quimioterapia postrasplante y TMO76. Aunque la supervivencia a 1 año en algunas series es cercana al 80%, el dificultoso manejo de la inmunosupresión durante el TMO73, así como otras complicaciones, nos hace recomendar este abordaje como una terapia a realizar sólo en centros con experiencia.
En el caso de las amiloidosis familiares, si se plantea la realización de TxC, este tendrá que ir asociado a la realización de un TxH que podrá ser simultáneo o secuencial al TxC73, 77, 78. Aunque se ha señalado que los depósitos cardiacos de amiloide mutado podrían actuar como «captadores» de proteína nativa79, creemos que no existe suficiente evidencia para sustentar en esto la necesidad de realizar trasplante simultáneo de hígado y corazón, lo que evidentemente añade un riesgo significativo y prácticamente el único beneficio que aporta es logístico.
En 2009 se publicó la experiencia española en el TxC de pacientes con amiloidosis entre 1984 y 200878. Durante ese periodo recibieron trasplante por amiloidosis 25 pacientes (el 0,4% del total de TxC) de los que 13 sufrían amiloidosis AL; 10, amiloidosis hereditarias por TTR y 2, amiloidosis AA. De los 10 pacientes con ATTR, 5 fueron sometidos a TxH, 3 fallecieron antes de recibirlo y 2 no se sometieron a él. La mortalidad aguda (primer mes post-TxC) de los pacientes con amiloidosis no fue diferente de la del grupo general, pero la supervivencia crónica era sensiblemente menor (el 36 frente al 64% a los 5 años)78. Por subgrupos, los pacientes con ATTR fueron los que mejor pronóstico mostraron, aunque probablemente esto está condicionado a que sólo 3 pacientes con amiloidosis AL se sometieron a TMO78.
Nuevas terapiasVarias moléculas que actúan sobre los depósitos de amiloide se han presentado en los últimos años como potenciales tratamientos para estos pacientes80. Algunas actúan eliminando el componente SAP de los depósitos de amiloide con el fin de que esto reduzca los agregados de amiloide y facilite su eliminación81. Estudios para evaluar la dosis y la tolerabilidad de alguna de estas moléculas están en marcha actualmente.
Otro abordaje prometedor es la estabilización de la molécula de TTR para evitar su pliegue anormal y su depósito mediante la administración de diflunisal o ácido flumenámico (antiinflamatorios que estabilizan la forma tetramérica de TTR) o nuevas moléculas (Fx-1006A). Un ensayo internacional sobre diflunisal (http://clinicaltrials.gov/show/NCT00294671) está ahora en marcha para pacientes con amiloidosis familiar TTR, pero sólo los pacientes con afección neuropática y sin afección cardiaca significativa son incluibles. Varios ensayos que evalúan la efectividad de Fx-1006A en pacientes con afectación neuropática o cardiaca (sólo pacientes con la mutación Val122Ile o la forma senil) se han completado recientemente. Los resultados todavía no han sido publicados.
CONCLUSIONESLa amiloidosis cardiaca es una entidad que agrupa en sí diversas enfermedades en función del subtipo del precursor amiloidótico. Puede aparecer aislada o, como es más frecuente, en asociación a afección de otros órganos y sistemas. Dado que el cardiólogo puede ser el primer profesional que se enfrente a esta enfermedad, debe conocer los distintos subtipos de amiloidosis que pueden producir afección cardiaca y plantear el diagnóstico de esta entidad ante todo paciente con clínica compatible.
Aunque ningún test no invasivo es 100% diagnóstico en esta enfermedad, la combinación de una historia familiar o clínica (cardiaca y/o extracardiaca) y los hallazgos ecocardiográficos y alteraciones en el ECG compatibles hacen de este diagnóstico una opción muy probable.
El diagnóstico precoz es fundamental en algunas formas de amiloidosis (AL y AA), ya que su tratamiento puede detener e incluso revertir el desarrollo de la enfermedad. La correcta identificación de las formas hereditarias de amiloidosis también es clave para planificar un tratamiento adecuado y una correcta evaluación de los familiares.
Actualmente se está investigando en nuevas terapias destinadas a estabilizar los precursores amiloideos y eliminar los depósitos.
CONFLICTO DE INTERESESNinguno
Autor para correspondencia: Unidad de Miocardiopatías, Sección de Insuficiencia Cardiaca y Trasplante, Servicio de Cardiología, Hospital Universitario Puerta de Hierro, Manuel de Falla 2, 28222 Majadahonda, Madrid, España. pablogpavia@yahoo.es