Palabras clave
INTRODUCCIÓN
La cardiopatía isquémica y la miocardiopatía delventrículo izquierdo se han considerado clásicamente la principal causa de las arritmias ventricu-lares (AV) y la muerte súbita cardiaca (MSC). Sinembargo, las arritmias originadas en el ventrículoderecho (VD) han atraído la atención de los científicos en las últimas dos décadas por diversas razones. Las AV originadas en el VD suelen afectar apacientes de menor edad y pueden conducir a laMSC. El mecanismo fisiopatológico de esas arritmias no se ha aclarado por completo y deja margenpara una investigación activa y diferentes interpretaciones. Por otro lado, el intrigante mundo de lagenética contribuye de manera creciente a explicarlos aspectos patogénicos, diagnósticos y pronósticos de estas arritmias.
MIOCARDIOPATÍA/DISPLASIAARRITMOGÉNICA DEL VENTRÍCULODERECHO
La miocardiopatía/displasia arritmogénica delventrículo derecho (DAVD) es una miocardiopatíacaracterizada por AV y anomalías estructurales progresivas del VD1,2. La degeneración miocárdicapuede extenderse al ventrículo izquierdo, sobre todoen las fases avanzadas de la enfermedad3. La DAVDpuede darse en formas esporádicas y familiares. Laenfermedad se caracteriza por una sustitución progresiva parcial o masiva del miocardio por tejidoadiposo o fibroadiposo. Esta infiltración constituyeun sustrato para la inestabilidad eléctrica y lleva aAV que van desde las extrasístoles ventriculares(EV) aisladas hasta las taquicardias ventriculares(TV) sostenidas o la fibrilación ventricular3,4. La prevalencia de la enfermedad en la población general se ha estimado en valores que van de 1/2.000 a1/10.000. El 80% de los casos se diagnostica en pacientes de edad inferior a 40 años. Debe sospecharseuna DAVD en todos los pacientes jóvenes con uncorazón aparentemente normal que sufren un sín-cope, TV o parada cardiaca. Como se comenta másadelante, la enfermedad debe sospecharse tambiénen deportistas que presentan síntomas compatiblescon arritmias (palpitaciones, síncope).
Aspectos genéticos
La DAVD es un trastorno heredable5. Hay unaclara incidencia familiar (un 30-50% de los casos) con un patrón de transmisión autosómico dominante, diversos grados de penetración y una expresión fenotípica polimórfica. Se ha descrito tambiénuna forma autosómica recesiva de la DAVD. Seasocia a una queratodermia palmoplantar y a unpelo lanudo (enfermedad de Naxos)6. Este tipo deDAVD lo causa una mutación en el gen de la placoglobina, cuyo producto es un componente de losdesmosomas y las uniones celulares de tipo adherens7. La primera mutación causante de unaDAVD no sindrómica fue descrita por Rampazzoet al8 en 2002. Dicha mutación se identificó en elgen de la desmoplaquina, que codifica un componente del desmosoma. En 2004, Gerull et al9 describieron 25 mutaciones del gen desmosómico cardiaco de la placofilina 2. Posteriormente seidentificaron otras mutaciones presuntamente causantes de enfermedad en la placoglobina y la desmoplaquina, así como en otros genes desmosómicos(los de la desmocolina 2 y la desmogleína 2) en pacientes con DAVD no sindrómica10. Actualmente seconsidera que la disfunción desmosómica es la víafinal común en la patogenia de la DAVD.
Se han localizado en el mapa cromosómico diferentes variantes genéticas de DAVD y se han descrito más de 140 mutaciones de DAVD causantesde enfermedad, la mayoría de ellas correspondientesa genes que codifican proteínas desmosómicas. Algunos genes no desmosómicos se han asociado también a una DAVD autosómica dominante, entreellos el gen del factor de crecimiento transformador β-3 (TGFβ3)11, que modula la expresión de las proteínas de contacto celular y el gen del receptor derianodina 2 (RyR2)12. El gen RyR2, que se describió primero en ocho familias, codifica receptoresque intervienen en la liberación del calcio del retículo sarcoplásmico. Sin embargo, sigue habiendoopiniones divergentes en cuanto a si debe considerarse o no que los pacientes con mutaciones del RyR2 sufren una DAVD o una TV polimórfica catecolaminérgica. La mutación que se ha descrito deforma más reciente afecta al gen TMEM43 y causauna variante de DAVD de penetración completa ymuy letal (DAVD5)13.
Desmosomas y DAVD
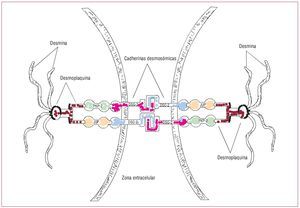
La integridad estructural y funcional del tejidocardiaco se basa en los desmosomas, las uniones tipo adherens (uniones adherentes) y las uniones tipo gap (uniones estrechas) situadas en los discos intercalares(fig. 1). La integridad de los desmosomas es necesaria para mantener la función normal de las unionesestrechas como canales intercelulares encargados delacoplamiento eléctrico y los mecanismos de señalización en la regulación del crecimiento, la diferenciación y el desarrollo celulares.

Fig. 1. En los desmosomas, las proteínas de la unión placoglobina (JUP), placofilina (PKP) 2 y desmoplaquina (DSP) anclan los filamentos intermedios de desmina a las cadherinas desmosómicas, con lo que aseguran la adhesión mecánica entre célula y célula. Las cadherinas desmosómicas, desmogleína (DSG) 2y desmocolina (DSC) 2, son proteínas transmembranarias que forman un dímero extracelular en forma de cremallera con la parte correspondiente de lascadherinas desmosómicas de la célula miocárdica adyacente. Modificado de Sen-Chowdhry et al14.
Las mutaciones genéticas causa de la DAVD danlugar a una haploinsuficiencia y una reducción dela expresión de las proteínas desmosómicas, quepueden predisponer a la rotura de los contactos celulares mecánicos, que puede desencadenarse poruna tensión mecánica del VD (como ocurre duranteel ejercicio o la actividad deportiva). La degeneración y la muerte de los miocardiocitos son la consecuencia anatomopatológica de estas mutaciones deproteínas de adhesión, con la consiguiente sustitución progresiva por tejido adiposo y fibroadiposo.
Anomalías estructurales
La DAVD se caracteriza por una sustitución delmiocardio por tejido adiposo y fibroso que inicialmente afecta al epicardio y más tarde al endocardio.Este proceso afecta la mayor parte de las veces a lasáreas posterior e inferior del tracto de entrada delVD adyacente a la válvula tricúspide, pero tambiénafecta al infundíbulo anterior y al vértice cardiaco,formando lo que se denomina «triángulo de ladisplasia»5. La pérdida de miocardio puede causar un adelgazamiento focal de la pared libre ventricular, con abombamiento focal de la pared del VDen la diástole y agrandamiento del tracto de salidaventricular derecho (TSVD). Desde el punto devista funcional, la enfermedad puede causar anomalías de la contracción generales o regionales, alteraciones de la función sistólica/diastólica del VD,formación de aneurismas del VD y dilatación e hipocinesia del VD15. El tabique interventricular sueleestar preservado. Por consiguiente, las biopsias endomiocárdicas, que generalmente se obtienen deltabique, pueden no ser diagnósticas. La biopsia endomiocárdica realizada en el VD tiene una sensibilidad baja, debido al carácter segmentario de la infiltración grasa. Las muestras deben obtenerse de lapared libre del VD, puesto que la sustitución fibroadiposa suele ser transmural y, por lo tanto, detectable con un abordaje endocárdico. No debesubestimarse el riesgo de perforación durante labiopsia endocárdica del VD en el vértice cardiaco yla pared libre del VD.
La DAVD debe diferenciarse de la enfermedadde Uhl, un trastorno congénito muy poco frecuente con ausencia de miocardio ventricular derecho, conlo que la pared del VD es delgada como el papel15.
Forma de presentación clínica
La DAVD se manifiesta generalmente en formade episodios de TV, con morfología de bloqueo derama izquierda del haz de His (BRIHH) y origen enel VD en adolescentes o adultos jóvenes aparentemente sanos. Las AV pueden ser asintomáticas ydetectarse en un ECG sistemático o pueden causarpalpitaciones, síncope o MSC. Se ha estimado quela DAVD explica hasta un 5-20% de los casos deMSC en individuos de menos de 35 años de edad15.
Sin embargo, las manifestaciones clínicas de la enfermedad pueden variar ampliamente en cuanto almomento de inicio y la gravedad. La interacciónentre diferentes sustratos genéticos con distintosgrados de penetración y la presencia de desencadenantes externos (como el ejercicio extenuante o elentrenamiento enérgico) explican este amplio espectro de formas de presentación clínica, que incluye palpitaciones, fatiga, dolor torácico atípico,síncope y MSC. La edad a la que se produce la primera manifestación oscila entre los 15 y los 35 años.El trastorno afecta a varones con mayor frecuenciaque a mujeres, y suele manifestarse en ellos con unaexpresión más amplia de la enfermedad. La insuficiencia cardiaca sintomática es una manifestación poco común de la DAVD y la mayor parte de lasveces se produce en estadios avanzados de la enfermedad. Los pacientes con antecedentes prolongados de DAVD tienen afectado el ventrículo izquierdo y sufren síntomas clínicos de insuficienciacardiaca biventricular16.
Diagnóstico
Según el Task Force Report on ARVD de 1994publicado por McKenna et al17, el diagnóstico deDAVD se basa en la presencia de factores estructurales, histológicos, electrocardiográficos, arrítmicosy genéticos y en los antecedentes familiares(tabla 1). Los pacientes deben cumplir dos criteriosmayores o un criterio mayor y dos criterios menoreso cuatro criterios menores para que se los considereafectados por una DAVD17. En 2002 se propusouna modificación de los criterios de la Task Forcepara el diagnóstico de la DAVD, en un caso de familiares de primer grado de un caso inicial. En estasituación, la presencia de una inversión de la ondaT precordial derecha (V2-V3), potenciales tardíosen el ECG de promediación de señal o TV con morfología de BRIHH o la observación de cambiosfuncionales o morfológicos leves del VD en las exploraciones de diagnóstico por la imagen debenconsiderarse criterios mayores con valor diagnóstico para la DAVD familiar18. Muy recientemente se ha publicado una nueva modificación de los criterios diagnósticos19 con la finalidad de aumentar lasensibilidad diagnóstica mediante el uso de las modalidades diagnósticas emergentes, los avances en lagenética de la DAVD y la introducción de parámetros cuantitativos (tabla 2). Así pues, la evaluaciónhabitual debe incluir la ecocardiografía bidimensional, electrocardiografía, electrocardiografía depromediación de señal, monitorización Holter de24 h, prueba de esfuerzo y antecedentes familiares.Cuando los resultados no sean concluyentes, sedebe considerar el uso de otras exploraciones deimagen y una biopsia endomiocárdica.


Anomalías electrocardiográficas
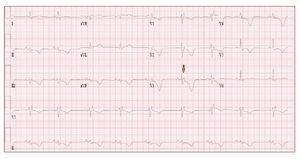
El ECG de los pacientes con DAVD suele mostrar un ritmo sinusal regular, con una duración delQRS > 110 ms en la derivación V1. Las alteracioneselectrocardiográficas incluyen ondas T invertidasen las derivaciones precordiales derechas más alláde V1, sin que haya bloqueo de rama derecha delhaz de His (BRDHH) y potenciales tardíos ventriculares derechos en forma de «ondas épsilon» en lasderivaciones V1-V3 (fig. 2).

Fig. 2. Electrocardiograma de un varón de21 años con una displasia arritmogénicadel ventrículo derecho. Se observa un bloqueo de rama derecha del haz de His, unaonda épsilon (flecha) y una inversión de laonda T difusa.
La inversión de la onda T en las derivacionesV1-V3 es una característica bien conocida del ECGen la DAVD y, en ausencia de BRDHH, de hechose ha propuesto que constituye un criterio diagnóstico mayor19. El patrón juvenil de la inversión de laonda T en V1-V3 o más allá es una variante normalen los niños de menos de 12 años de edad. Esta variante está presente en un 1-3% de la población sanade 19 a 45 años de edad, pero se da en el 87% de lospacientes con DAVD. Las ondas épsilon son potenciales eléctricos de «postexcitación», de pequeñaamplitud, que se producen en el segmento ST tras elfinal del complejo QRS. Estas ondas, que se observan en el 33% de los pacientes con DAVD, seconsideran un criterio diagnóstico mayor para la DAVD. Puede haber diferentes anomalías del ECGen función de cuál sea la extensión de la enfermedad. Sin embargo, la presencia de un ECGnormal no debe hacer que se descarte inicialmenteel diagnóstico de DAVD20.
Diagnóstico por la imagen
Las técnicas de diagnóstico por la imagen utilizadas para diagnosticar anomalías morfofuncionalescompatibles con una DAVD son la angiografía convencional, la ecocardiografía, la tomografía computarizada (TC), la angiografía radioisotópica y laresonancia magnética (RM). La angiografía ventricular derecha se ha considerado históricamente lamejor exploración de imagen para el diagnóstico dela DAVD y se ha demostrado que tiene una elevadaespecificidad (90%)21. La ecocardiografía es menosinvasiva y un método de primera línea para evaluara los pacientes en los que se sospecha una DAVD oen el examen de detección sistemático de familiares.La RM permite diferenciar la grasa del músculo.Además, la RM permite efectuar una evaluacióncuantitativa y muy exacta del tamaño y la funcióndel VD. Aunque la utilidad de la RM en el diagnóstico de la DAVD está ampliamente aceptada, estatécnica conduce a veces a un sobrediagnóstico deDAVD. La sensibilidad y la especificidad de la detección mediante RM de la grasa intramiocárdicadel VD en el diagnóstico de la DAVD es variable yoscila entre el 22 y el 100%22-25. La identificación dela grasa puede ser difícil, ya que el VD es una estructura delgada y las áreas de miocardio afectadaspueden ser muy pequeñas. Además, actualmente esbien sabido que la presencia de grasa en el miocardiodel VD puede ser normal. Diferenciar una infiltración adiposa patológica en áreas en que normal-mente está presente la grasa epicárdica adyacente,como el surco auriculoventricular y la parte anteroapical del VD, puede ser especialmente difícil.
También se han descrito zonas aisladas de sustitución grasa en pacientes ancianos, con el uso prolongado de corticoides, en la obesidad, en otras miocardiopatías y en la TV-TSVD idiopática26. Así pues,los médicos deben evitar el diagnóstico de DAVDcuando las únicas anomalías estructurales son lasobservadas en la RM.
La RM con contraste tardío de gadolinio puedelocalizar alteraciones fibroadiposas en el miocardiodel VD. La variedad de DAVD fibroadiposa es másfrecuente que la DAVD adiposa. Así pues, la RMcon contraste tardío de gadolinio puede tener unpapel gracias a su capacidad de detectar casos iniciales y sutiles de DAVD que de otro modo seríandiagnosticados de forma errónea27,28. Se ha señaladoque demostrar la presencia de tejido fibroso tienemayor importancia diagnóstica que la observaciónde grasa sola29.
Mapas de voltaje: un instrumento diagnósticoimportante
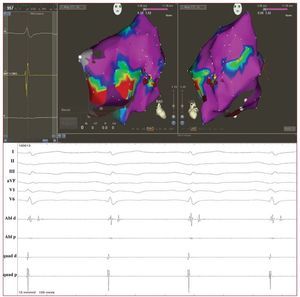
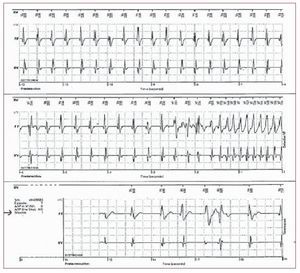
Están apareciendo datos que indican un papelcreciente de los mapas de voltaje endocárdicos paraidentificar la presencia de una cicatriz en el VD enlas fases iniciales de la enfermedad. La técnica tiene un potencial de identificación exacta de la presencia, la localización y la extensión del sustratoanatomopatológico subyacente en la DAVD, mediante la demostración de regiones del VD de bajovoltaje, es decir, de cicatrices electroanatómicas(fig. 3). En los pacientes con DAVD, se ha demostrado que las cicatrices del VD identificadas mediante mapas electroanatómicos corresponden aáreas de depleción miocárdica y están correlacionadas con los signos histopatológicos de atrofiamiocárdica y sustitución fibroadiposa en la biopsiaendomiocárdica30. Corrado et al demostraron quelos mapas de voltaje pueden ser útiles en el diagnóstico diferencial entre la TV-TSVD y la forma temprana de la DAVD. De hecho, en los pacientes conAV originadas en el VD y con un VD aparentemente normal en las exploraciones de diagnósticopor la imagen convencionales, la presencia de unárea de regiones de bajo voltaje en el TSVD identificó un subgrupo con alto riesgo de recurrencia dela TV y MSC durante el seguimiento clínico31. Muyrecientemente, Wijnmaalen et al han demostradoque los pacientes con TV derecha asociada a la presencia de cicatrices, con o sin un diagnóstico deDAVD establecido según los criterios de la TaskForce, tenían una tasa de recurrencia de TV superior a la de los pacientes sin cicatrices electroanatómicas en el VD32. Todos estos datos confirman quelos mapas de voltaje podrían ser un instrumentodiagnóstico importante con implicaciones pronósticas y terapéuticas.

Fig. 3. Arriba: reconstrucción con mapade voltaje del ventrículo derecho en ritmosinusal utilizando un sistema de elaboración de mapas electroanatómicos (Carto3, Biosense Webster, Estados Unidos)en un varón de 43 años con una formamanifiesta de displasia arritmogénicadel ventrículo derecho y una taquicardiaventricular monomórfica sintomática. Seidentifican áreas de cicatrización (voltajelocal < 1,5 mV) en la pared anterolateral del ventrículo derecho (parte basal).Los puntos rojos corresponden a la líneade ablación de radiofrecuencia aplicadacon la finalidad de bloquear el circuito demacrorreentrada asociado a la cicatriz.Abajo: potenciales tardíos endocavitariosregistrados con el catéter de elaboraciónde mapa en las regiones de la cicatriz ysu entorno.
Diagnóstico diferencial
El diagnóstico diferencial principal de la DAVDes el que debe hacerse con la TV-TSVD, la sarcoidosis, la miocardiopatía dilatada idiopática y lamiocarditis aislada. Tanto la TV-TSVD como laDAVD se producen en individuos jóvenes aparentemente sanos, y ambas pueden manifestarse porEV o TV con un BRIHH y un eje inferior. Aunqueno es difícil diagnosticar un caso manifiesto deDAVD, la diferenciación de ésta en sus fases iniciales respecto a la taquicardia del TSVD, un trastorno arrítmico generalmente benigno y que notiene carácter familiar, continúa siendo un verdadero reto clínico. Cuando las exploraciones tradicionales (ECG, Holter) y las técnicas de diagnósticopor la imagen (ecocardiografía, angiografía ventricular derecha, RM) continúan dejando dudas diagnósticas, los mapas de voltaje del VD parecen serun nuevo instrumento útil para diferenciar las dosentidades.
Papel del análisis genético
Una aplicación clínica clave del análisis genéticoes la realización de pruebas de confirmación de loscasos iniciales con objeto de facilitar la interpretación de los resultados limítrofes y el examen de detección en cascada de los familiares14,33. La utilidadde las determinaciones del genotipo de los familiares antes de que manifiesten un fenotipo clínicomaligno puede resultar crucial en la prevención dela MSC. El tratamiento de los familiares con formasno manifiestas de DAVD diagnosticadas medianteun análisis de genética molecular puede prevenir laMSC en estos individuos, mediante un seguimientoclínico seriado (ECG, ecocardiografía, Holter de24 h, identificación temprana de los síntomas), mediante modificaciones del estilo de vida (limitaciónde la actividad física extrema) y mediante un tratamiento profiláctico en caso necesario (fármacos antiarrítmicos, desfibriladores cardiacos automáticosimplantables [DAI]). A pesar de su fiabilidad, la secuenciación genética es un proceso laborioso y decostes elevados, sobre todo en el estudio de laDAVD, en la que el número de genes involucradospuede ser elevado.
Muy recientemente, se ha descrito una nuevaprueba diagnóstica basada en el análisis inmunohistoquímico desmosómico de muestras miocárdicashumanas obtenidas mediante biopsia endomiocárdica en una población de pacientes pequeña34. Eseestudio indicó que el nivel de señal inmunorreactivapara la proteína desmosómica placoglobulina estáreducido en los discos intercalares de los pacientescon DAVD. Es interesante señalar que los nivelesde señal de placoglobulina estaban reducidos nosólo en el miocardio ventricular derecho, con alteraciones anatomopatológicas características de unasustitución fibroadiposa, sino también en el ventrículo izquierdo y el tabique interventricular de aspecto normal. Además, esto no se observó en otrasformas de enfermedad del músculo cardiaco.
Estratificación del riesgo
Los datos existentes indican que la disfunción delVD grave, la afección del ventrículo izquierdo, elsíncope, la edad temprana, el sexo masculino, losantecedentes de parada cardiaca, la TV rápida ymal tolerada con diferentes morfologías, y la incidencia familiar de muertes súbitas juveniles son losprincipales factores que determinan una malaevolución35,36. Los pacientes de alto riesgo presentansignos clínicos de insuficiencia cardiaca derecha y/otienen una disfunción ventricular izquierda, así como antecedentes de TV36. Estos pacientes debenser considerados candidatos a un manejo terapéutico enérgico. En cambio, los pacientes sin TVtienen un riesgo muy bajo de sufrir episodios cardiacos. Continúa habiendo debate respecto a si elestudio electrofisiológico puede ser útil37 o no38,39 para predecir la aparición de una TV durante el seguimiento. Las recomendaciones de estilo de vida,es decir, la limitación de la actividad física enérgica,pueden mejorar la evolución a largo plazo.
Tratamiento
El objetivo principal de la estrategia terapéuticaes la prevención de la MSC. Los tres principalestratamientos de que disponemos para los pacientescon DAVD son los fármacos antiarrítmicos, laablación por catéter y el uso de DAI.
Fármacos antiarrítmicos
Los pacientes con DAVD y sin antecedentes desíncope o parada cardiaca, que presentan EV, EVen parejas o salvas ventriculares cortas, no suelentener un aumento del riesgo arrítmico y, por consiguiente, no requieren un tratamiento antiarrítmicoespecífico. En los pacientes con TV sostenida, eltratamiento con fármacos antiarrítmicos tiene comoobjetivo no tanto la supresión de las recurrencias dela TV, sino principalmente la prevención de laMSC. Sotalol, a dosis de 320-480 mg/día, ha sido identificado como el fármaco que proporciona mejores resultados, con una tasa general de eficacia del68%40. Es interesante señalar que un reciente estudio ha indicado que, en una cohorte de pacientescon DAVD caracterizados de forma rigurosa, losbloqueadores beta no tuvieron efectos nocivos niprotectores contra las AV clínicamente relevantes;el estudio puso de manifiesto también que el sotalolno era eficaz y que la amiodarona era el fármacocon una mayor eficacia41. Los fármacos antiarrítmicos de clase I resultaron eficaces tan sólo en unaminoría de pacientes con DAVD. Hasta la fecha nose han realizado estudios prospectivos y aleatorizados sobre la eficacia de los fármacos antiarrítmicos en la DAVD.
Ablación con catéter
Las indicaciones actuales para la ablación por catéter en pacientes con DAVD son la TV monomórfica y bien tolerada con formas localizadas de la enfermedad y refractariedad a la medicación o la TVincesante o con descargas frecuentes de DAI. Eneste último caso, la ablación por catéter puede desempeñar un papel importante como opción de tratamiento paliativo o adyuvante para la reducción ola supresión de la TV.
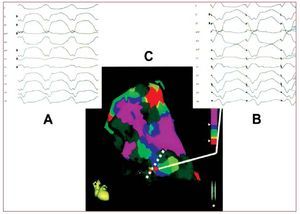
La TV en la DAVD es el resultado de un circuitode reentrada relacionado con una cicatriz, de manera similar a lo observado en un contexto tras infarto de miocardio. La ablación por catéter conmapas de voltaje del VD, mediante el empleo detécnicas de elaboración de mapas convencionales oelectroanatómicas, puede aportar unos resultados a corto plazo favorables (fig. 4)42. Sin embargo, comoconsecuencia de la progresión de la enfermedad, lasrecurrencias de AV a partir de nuevos sustratosarritmogénicos son frecuentes. Wichter et al hanpresentado su experiencia con la ablación por catéter en 30 pacientes con DAVD. Se alcanzaronunos resultados a corto plazo favorables, con supresión completa de la TV refractaria a la medicación, en 22 pacientes (73%). Sin embargo, los resultados a largo plazo fueron menos satisfactorios, detal manera que 18 pacientes (60%) presentaron recurrencias de la TV. Es de destacar que la mayoríade las recurrencias tardías (más de 1 año) de la TVobservadas en este estudio mostraron una morfología del complejo QRS diferente que en la TV quehabía motivado el tratamiento, lo cual indica quepueden aparecer nuevos sustratos arritmogénicosdurante la progresión de la enfermedad a largoplazo en su evolución crónica43. Otros autores hanpresentado iguales resultados44,45. También se haconsiderado la posibilidad de un sustrato subepicárdico para las recurrencias de la TV45.

Fig. 4. Ejemplo del uso combinado demapas de voltaje y de ritmo de marcapasos en un paciente con displasia arritmogénica de ventrículo derecho. A: electrocardiograma de 12 derivaciones con lataquicardia ventricular clínica. B: electro-cardiograma de 12 derivaciones durantela elaboración del mapa de ritmo de marcapasos con la punta del catéter (puntorojo en los lugares situados a lo largo delborde de la cicatriz, panel C), que muestrauna morfología electrocardiográfica casiidéntica a la de la taquicardia ventricularclínica, lo cual indica que este lugar es lazona de salida que puede ser crítica de uncircuito de macrorreentrada. Modificadocon permiso de Arruda et al42.
Desfibrilador cardiaco automáticoimplantable
En los pacientes con DAVD, el tratamiento conDAI mejora el pronóstico a largo plazo y la super-vivencia si se aplica a una población de alto riesgoseleccionada y como prevención secundaria(fig. 5)45-47. Sin embargo, los criterios para la selección óptima de los pacientes en los que se obtieneun efecto beneficioso con la implantación de un DAI para la prevención primaria no se han definidoaún. En el futuro, cabe prever un papel más importante de la genética en la toma de decisiones46,47.

Fig. 5. Un electrograma intracardiaco registrado en un desfibrilador automáticoimplantable que muestra una taquicardiaventricular rápida con la consiguientedesfibrilación apropiada en un varón jovencon displasia arritmogénica de ventrículoderecho.
Es de destacar que la colocación de un DAI enpacientes con DAVD puede conducir a diversascomplicaciones de manera más frecuente que enotras enfermedades en las que es necesario el uso deDAI. Una complicación frecuente es la debida a laprogresión de la atrofia miocárdica y la posteriorsustitución por grasa en el lugar de la implantacióndel electrodo, lo cual da lugar a una pérdida de lafunción de percepción del electrodo de desfibrilación del VD, que hace necesario sustituir dicho electrodo. Así pues, la indicación para el tratamientocon DAI en la DAVD debe ponderar los posiblesefectos beneficiosos en comparación con el riesgode complicaciones.
Cuando la enfermedad ha progresado a una insuficiencia ventricular derecha o biventricular, sedebe aplicar el tratamiento indicado actualmentepara la insuficiencia cardiaca, que incluye diuréticos, bloqueadores beta, inhibidores de la enzimade conversión de angiotensina y anticoagulantes.En caso de insuficiencia cardiaca derecha refractaria, el trasplante de corazón puede ser la únicaalternativa.
SÍNDROME DE BRUGADA
Introducción
El síndrome de Brugada (SB) es una enfermedadgenética que causa arritmias cardiacas y se caracteriza por la aparición de MSC en individuos jóvenessin ninguna evidencia de cardiopatía estructural.Dada la ausencia macroscópica de anomalías cardiacas estructurales, el SB se clasifica como una«enfermedad eléctrica primaria» o canalopatía cardiaca. El SB está relacionado con anomalías estructurales y funcionales del canal del sodio. Diversasobservaciones fisiopatológicas indican que los trastornos eléctricos relacionados con el SB se sitúanprincipalmente en el VD y en especial en el TSVD.
Los pacientes con SB presentan un patrón deECG que se caracteriza por una elevación del segmento ST en las derivaciones precordiales derechas(V1-V3)48 y un BRDHH incompleto o completo. LaMSC se produce por una TV polimórfica y/o por fibrilación ventricular (FV)49. Se cree que el SB es lacausa de un 4-12% del total de MSC y de hasta un20% de las MSC en individuos que no presentancardiopatías estructurales50,51. Dado que el ECG esdinámico y a menudo la alteración está oculta, resulta difícil estimar la prevalencia real de la enfermedad en la población general51,52. La prevalenciadel SB se estima en 1-5/10.000 habitantes en todo elmundo. La frecuencia es menor en los países occidentales y es más alta (> 5/10.000) en el sudesteasiático, sobre todo en Tailandia y Filipinas, dondeel SB se considera una causa importante de muertenatural de personas jóvenes53,54.
Aspectos genéticos
La primera mutación relacionada con este síndrome fue descrita en 1998 por Chen et al55 y se identificó en el gen SCN5A que codifica la subunidadalfa del canal de sodio cardiaco55-57. Las mutacionesdel gen SCN5A se identifican en la actualidad en un18-30% de los pacientes con SB. La transmisión hereditaria en el SB se produce a través de un modo detransmisión autosómico dominante. Hasta la fecha,se han hallado otras 293 mutaciones diferentes en elmismo gen. En 2002, Weiss et al describieron un segundo locus en el cromosoma 3, que no estaba ligadoal SCN5A: el gen identificado fue el gen de tipo glicerol-3-fosfato deshidrogenasa 1 (GPD-1L)58. Se hademostrado que las mutaciones del gen GPD-1L, que codifican una enzima que regula el tráfico por elcanal de sodio cardiaco hacia la superficie celular, reduce las corrientes de entrada de sodio en aproximadamente un 50%59. Recientemente se ha demostradotambién que ciertas mutaciones de los genes CACNA1c y CACNB2b60, que codifican canales delcalcio, y mutaciones del gen KCNE361, que codificauna subunidad reguladora de la corriente Ito de potasio, producen un fenotipo de SB.
El ECG del SB no es el único fenotipo ligado amutaciones del gen SCN5A62. La evidencia recienteindica que hay un considerable solapamiento de laforma de presentación clínica («síndromes solapados»). El solapamiento registrado con más frecuencia es la concomitancia de SB y enfermedad deconducción cardiaca (enfermedad de Lev-Lenegre,síndrome del nódulo sinusal enfermo). El solapamiento entre los fenotipos LQT3 y SB se ha descritotambién en varios casos63. En los últimos años se hadescrito también la asociación de la fibrilación auricular con canalopatías de sodio conocidas, como elSB, la enfermedad de conducción cardiaca progre-siva, los síndromes de QT corto y el LQT364-66. Es interesante señalar que se ha demostrado que la fibrilación auricular puede ser la primera expresiónfenotípica de una forma latente del SB que se manifiesta únicamente años después67.
Fisiopatología
Se han propuesto dos hipótesis principales paraexplicar el mecanismo fisiopatológico de las anomalías del ECG y la propensión a las AV en los pacientes con SB51. En 1999 se propuso la teoría del«deterioro de la repolarización»68. Esta teoría se basaba en una expresión no homogénea de la corriente transitoria de salida de potasio (Ito) entre elepicardio y el endocardio. La Ito, que origina unafase de repolarización temprana durante el potencial de acción (PA), se expresa en mayor medida enel epicardio. En presencia de pérdida o reducciónde la función de los canales de sodio, aparece unaforma de PA de «pico y cúpula». El efecto del potencial de acción será más evidente en la capa epicárdica, en la que la fuerza de la Ito es mayor. Asípues, una discrepancia en la forma del PA entre endocardio y epicardio dará lugar a los patrones delECG de superficie del SB. Las anomalías en el ECGde superficie serán proporcionales a la discrepanciaen el PA, dando lugar a alteraciones leves del ECG(elevación del ST en silla de montar, punto J< 0,2 mV) o alteraciones notables (tipo convexo o cove-type, punto J > 0,2 mV) según el grado en elque se haya dañado el canal del sodio y la diferenteexpresión de la corriente Ito. La conducción de lacúpula del PA de los lugares en que se mantiene alos lugares en que se pierde produce una reexcitación local a través de un mecanismo de reentradade fase 2 con una extrasístole directamente acoplada que se produce durante el periodo vulnerable.Estos EV pueden desencadenar entonces AV malignas.
La segunda teoría, denominada «teoría de ladespolarización»69, se basa en la presencia de un retraso en la conducción en el TSVD. Según estateoría, la forma del PA se mantiene pero el PA delTSVD se retrasa respecto al PA del VD. Estas diferencias temporales del PA en áreas adyacentes delmiocardio pueden ser origen de circuitos de reentrada desencadenados por EV originados en la zonalimítrofe entre la despolarización temprana y latardía.
Una tercera teoría, basada en la expresiónanormal de la cresta neural en el desarrollo miocárdico del TSVD y las estructuras circundantes, hasido propuesta recientemente por Elizari et al70. Durante la embriogénesis, la cresta neural cardiacadesempeña un papel crucial en la morfogénesis delTSVD, que comprende la pared libre y el tabiqueaortopulmonar y los grandes vasos. Una poblaciónde células de la cresta neural cardiaca migra haciael polo arterial del corazón embrionario y facilita laproliferación de las células miocárdicas, su diferenciación y la miocardialización del TSVD. Por otraparte, una segunda vía de las células de la crestaneural cardiaca utiliza el polo venoso como entradaal corazón y es esencial en la formación del nóduloauriculoventricular, el haz de His, el inicio de lasramas del haz y el tejido auricular.
Por consiguiente, las anomalías en la formacióndel corazón derecho relacionadas con el origen extracardiaco de la cresta neural podrían explicartodas las anomalías eléctricas (arritmias supraventriculares, alteraciones de la conducción, AV) quepueden manifestarse en pacientes con SB y otrossíndromes solapados.
En particular, una expresión no homogénea deconexinas, que son moléculas con elevada expresiónen las células de la cresta neural, puede ser la causade la migración incorrecta de las células de la crestaneural hacia el corazón embrionario. El hecho deque la migración de las células de la cresta neural seproduzca en un momento inadecuado a causa deuna mala función de las conexinas puede tener unefecto nocivo en el remodelado del tejido cardiacodel VD. Así pues, la miocardialización anormal deTSVD que depende de la migración de las célulasde la cresta neural podría explicar las heterogeneidades de repolarización que subyacen al fenotipode SB. Este modelo plantea la hipótesis de una distribución desigual de las fuerzas de repolarización.Según esta teoría, los gradientes de repolarizaciónque causan una elevación del segmento ST se producen no sólo entre epicardio y endocardio (debidoa la mayor expresión de la Ito en el epicardio que enel endocardio), sino también entre el TSVD y las estructuras circundantes normales.
Forma de presentación clínica
Los pacientes con SB pueden presentar una amplia variedad de síntomas, que van del individuocompletamente asintomático al paciente que fallecede forma súbita. Se han descrito casos de síncope,crisis convulsivas, palpitaciones y respiración agónica nocturna como síntoma de presentación inicial. Hasta un 20% de los pacientes de los países occidentales y hasta un 30% de los de Japón presentantaquicardias supraventriculares concomitantes66, lamás frecuente de las cuales es la fibrilación auricular, que puede ser también la primera manifestación de la enfermedad67.
El SB se considera principalmente una enfermedad arritmogénica de los varones adultos (80%),y la media de edad en el momento de la muerte súbita es 40 años71. Los estudios clínicos indican quela hormona masculina testosterona puede ser origende las diferencias de prevalencia del SB entre lossexos72-74.
ECG y características clínicas
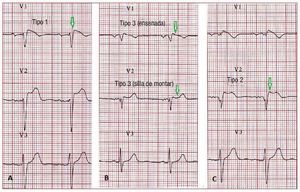
En la actualidad, se reconocen tres patrones derepolarización del ECG según la Second ConsensusConference on Brugada Syndrome (fig. 6). Eltipo 1, que es el único diagnóstico de SB, se caracteriza por una elevación del segmento ST en ensenada ≥ 2 mm (0,2 mV) seguido de una onda T negativa oplana. El patrón de repolarización del ECG detipo 2 se caracteriza por una elevación del segmentoST, que tiene un aspecto de «silla de montar», conun despegue alto en la elevación del segmento ST ≥ 2 mm y una onda T positiva o bifásica. El tipo 3presenta un aspecto de silla de montar o de ensenada, con una elevación del segmento ST < 1 mm.Los tipos 2 y 3 no son diagnósticos para el SB, peroel diagnóstico de SB se considera también positivocuando se observa un tipo 2 o 3 en más de una derivación precordial derecha en la situación basal y seproduce una conversión al patrón diagnóstico detipo 1 tras la administración de un bloqueador del canal del calcio75. El registro del ECG en las derivaciones V1 y V2 en espacios intercostales más altos(tercero y segundo) aumenta la sensibilidad y la especificidad del diagnóstico en la detección del fenotipo de Brugada76,77.

Fig. 6. A: electrocardiograma de un síndrome de Brugada de tipo 1 espontáneo.B: patrones de síndrome de Brugada detipo 3 en «silla de montar» y en «ensenada». C: patrones de síndrome de Brugadade tipo 2. Solamente el tipo 1 debe considerarse diagnóstico de un síndrome deBrugada.
Por consiguiente, según los criterios de la SecondConsensus Conference, el SB se diagnostica cuandose observa una elevación del segmento ST de tipo 1en más de una derivación precordial derecha(V1-V3) en presencia o en ausencia de un bloqueador del canal del sodio y conjuntamente conuno de los siguientes eventos: fibrilación ventriculardocumentada, TV polimórfica (autolimitada), antecedentes familiares de MSC antes de los 45 años,presencia de ECG de tipo ensenada en familiares,inducibilidad de TV con estimulación eléctrica programada o síncope. En los pacientes que presentanel patrón de ECG de tipo ensenada característicosin otros criterios clínicos, debe considerarse quepresentan un patrón de ECG de Brugada y no unSB. Muy recientemente, hemos demostrado que unECG de tipo 1 en una sola derivación precordial essuficiente para el diagnóstico78.
Los tres patrones que se han descrito pueden observarse de manera espontánea en registros de ECGseriados de un mismo paciente, al igual que una«seudonormalización» del ECG79. En la prácticaclínica, estas fluctuaciones del ECG pueden hacermuy difícil identificar a los individuos afectados porun SB y en riesgo de sufrir una MSC. Además delos bloqueadores de canales de sodio, se ha descritoque muchos agentes y trastornos hacen que se manifieste un fenotipo de ECG de SB tipo 1, entreellos la temperatura corporal, los cambios del tonodel sistema autónomo y los fármacos que afectan ala función de canales iónicos, como antagonistasdel calcio, bloqueadores beta, antiarrítmicos y psicotropos y la toxicidad del alcohol o la cocaína75.
Recientemente, Amin et al han mostrado que elejercicio, en especial durante la fase de recuperación, puede hacer que se manifieste un ECG de SBtipo 1 o aumente la elevación del punto J máximoprecordial80.
Instrumentos diagnósticos: exposicióna fármacos antiarrítmicos de clase I
Dado que el ECG es dinámico y, por lo tanto, lamarca distintiva del ECG que es característicapuede quedar oculta, se ha propuesto que la exposición a bloqueadores del canal del sodio que aumentan la disfunción de dicho canal puede ser uninstrumento útil para el diagnóstico del SB. En laactualidad, la ajmalina, administrada en una dosisintravenosa de 1 mg/kg en 5 min, constituye elagente de primera elección, dada sus elevadas sensibilidad y especificidad en la identificación de portadores génicos (el 80 y el 94,5% respectivamente)81. Además, la semivida corta y la duración breve desus efectos electrofisiológicos hacen que sea más seguro que otros fármacos antiarrítmicos.
Estratificación del riesgo
La presencia de una elevación espontánea delsegmento ST precordial derecho (de tipo ensenada),los antecedentes de síntomas clínicos (síncope,muerte súbita abortada) y el sexo masculino sonfactores de riesgo de aparición de episodios clínicosmalignos en los pacientes con SB49,82-84.
Hay acuerdo general respecto a que los pacientesque han sobrevivido a parada cardiaca tienen unriesgo elevado de recurrencia de episodios arrítmicos con peligro para la vida y que, por lo tanto,requieren el empleo de un DAI34,37. Los antecedentes de síncope pueden darse en hasta un 23% delos pacientes que sufren una parada cardiaca84. Elsíncope de mecanismo neurógeno se ha asociado recientemente al SB, pero no se conocen todavía susrepercusiones en cuanto al pronóstico y la estratificación del riesgo85. Se ha observado de manera uniforme una tendencia del sexo masculino a presentarmás episodios de arritmia en todos los estudios, yen un reciente metaanálisis se ha definido inclusoesta característica como un factor predictivo independiente de una peor evolución86. Continúa habiendo controversia respecto a cuál es el mejor enfoque terapéutico en los pacientes asintomáticos yrespecto a si el estudio electrofisiológico puede serútil o no para predecir una evolución adversa84,87-88.
Tratamiento
El único tratamiento que ha demostrado la prevención de la muerte súbita en pacientes con SBsintomáticos es el DAI50,89,90. Puede usarse quinidinacomo opción de tratamiento farmacológico. Su empleo podría resultar especialmente útil en pacientescon un DAI y múltiples descargas42 y como opciónterapéutica inicial en pacientes jóvenes con unriesgo elevado de arritmias malignas. Sin embargo,no disponemos todavía de datos científicos realessobre su eficacia.
TAQUICARDIA VENTRICULAR DE TRACTODE SALIDA VENTRICULAR DERECHO
Las TV-TSVD son la forma más frecuente de TVidiopáticas91,92. En la mayor parte de los casos (70-80%)93 la TV-TSVD se origina realmente en elTSVD. Son posibles también otros orígenes comoel tabique, el tracto de salida ventricular izquierdo,la arteria pulmonar, el seno de Valsalva aórtico, elárea próxima al haz de His y la superficie epicárdica de los ventrículos94. La morfología característica dela TV-TSVD es la de una taquicardia de complejoQRS ancho con morfología de BRIHH y un eje inferior (fig. 7). La TV es monomórfica y general-mente no es de carácter familiar.

Fig. 7. Este electrocardiograma de 12 derivaciones corresponde a una mujer de 31años que presentaba una taquicardia ventricular sostenida, originada en el tractode salida ventricular derecho (complejosQRS de tipo bloqueo de rama izquierdadel haz de His y eje inferior). Unas pocas aplicaciones de radiofrecuencia casi1 cm por debajo de la válvula pulmonaren el lado de la pared libre hicieron quela taquicardia ventricular dejara de serinducible.
La forma de presentación clínica de la TV-TSVDes variable, y los síntomas suelen aparecer entre latercera y la quinta décadas de la vida95. La TVTSVD se produce más frecuentemente en las mu-jeres96. Se conocen dos formas fenotípicas de la TVTSVD: la TV sostenida inducida por el esfuerzo ola tensión emocional y la TV monomórfica repetitiva no sostenida que aparece en reposo. Ambasformas se caracterizan por la sensibilidad a la adenosina. La TV no sostenida que se produce general-mente en forma de salvas repetitivas de TV monomórfica es frecuente y constituye un 60-92% de loscasos en las series publicadas. La mayor parte delos pacientes presentan palpitaciones o presíncope yexcepcionalmente pueden sufrir un síncope. La taquicardia se desencadena generalmente por el ejercicio o la tensión emocional. El ECG en reposo deestos pacientes no presenta anomalías identificables. La ecocardiografía y la angiografía coronariason normales en la mayoría de los pacientes97. LaRM puede mostrar anomalías en hasta un 70% delos pacientes, incluido el adelgazamiento focal, lareducción del grosor de la pared y el movimientoanormal de ésta98. Sin embargo, no se conoce el significado real de estas «anomalías».
La TV-TSVD muestra un curso benigno, lo cualindica que, en la mayoría de los casos, la arritmiano es una forma frustrada de una miocardiopatíaoculta. Gaita et al describieron a 61 pacientes conEV de TSVD, con los que se contactó 15 años después de la visita inicial; ninguno de los pacienteshabía fallecido por MSC ni sufrió una DAVD enese estudio99.
Sin embargo, algunos autores han indicado quelas «arritmias malignas» como la fibrilación ventricular y/o las TV polimórficas se asocian a una TVidiopática o a EV originadas en el TSVD100,101. Los pacientes que pueden tener un riesgo elevado depresentar esta variante «maligna» parecen ser losque tienen: a) antecedentes de síncope; b) una TVmuy rápida (frecuencia cardiaca > 230 lpm); c) EVcon un intervalo de acoplamiento corto, y d) unaduración media prolongada del QRS de la EV iniciadora que tiene su origen en el TSVD101. Clara-mente hay que considerar una ablación por catéteren los pacientes con estas últimas características.
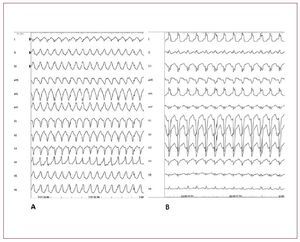
En el diagnóstico diferencial debe sospecharseclaramente una posible DAVD e investigarse supresencia. La primera característica que puede diferenciar la TV-TSVD de la DAVD es que la TVTSVD se manifiesta siempre con una morfología deBRIHH y eje inferior. La TV en la DAVD sueletener una morfología de BRIHH, pero el complejoQRS puede tener orientaciones diferentes en el ejefrontal. El diagnóstico diferencial de la TV-TSVDdebe descartar también la taquicardia asociada a fi-bras auriculofasciculares, la taquicardia de reentrada auriculoventricular a través de una vía accesoria del lado derecho y la TV aparecida tras lareparación de una tetralogía de Fallot (fig. 8).

Fig. 8. A: taquicardia ventricular (TV) sostenida originada en el tracto de salida ventricular derecho (TSVD). B: TV sostenida debida a fibras auriculofasciculares. Aunqueambas TV se manifiestan por un complejoQRS amplio y un bloqueo de rama izquierda del haz de His, el eje del QRS en el plano frontal difiere, y ello refleja la presenciade dos mecanismos y orígenes diferentes.En A, la TV tiene un origen focal y la masamiocárdica ventricular se activa de arribaabajo. En B, la TV depende de un circuitode reentrada y la masa miocárdica ventricular se activa en la dirección contraria, deabajo arriba, lo cual hace que la direccióndel eje del QRS en el plano frontal sea diferente. Las fibras auriculofascicularesson una forma de vía accesoria infrecuente pero muy bien definida. Estas víasaccesorias tienen su origen en la aurículaderecha, en el anillo tricuspídeo parietal,y terminan en la parte distal de la ramaderecha del haz o cerca de ella. Muestranuna conducción decremental lenta y exclusivamente anterógrada. Dado que estasvías se encuentran en el lado derecho, loscomplejos QRS preexcitados muestran unamorfología similar a la del bloqueo de ramaizquierda del haz de His.
Mecanismo de la TV-TSVD
La actividad desencadenada se considera el mecanismo subyacente en la TV-TSVD, debido a las posdespolarizaciones tardías que se producen através de la acción del adenosinmonofosfato cíclico (AMPc)102,103. En los pacientes que presentanuna TV-TSVD hay una sobrecarga de calcio intracelular. La sobrecarga de calcio citosólico potenciala función del intercambiador de Na+/Ca++ que dalugar a un aumento de la corriente de entrada y unretraso en la posdespolarización. La adenosina eseficaz para interrumpir la TV-TSVD gracias a sucapacidad de reducir el AMPc, que es crucial en laregulación del calcio intracelular. La taquicardiapuede ser inducible por extraestímulos programados o por una salva de latidos de marcapasosen el ventrículo o la aurícula o mediante la infusión de isoproterenol durante un estudio electrofisiológico.
Tratamiento
La estrategia terapéutica para la TV-TSVD incluye el uso de fármacos antiarrítmicos y la ablación por catéter. Si los síntomas son muy leves o infrecuentes, no es necesario ningún tratamiento. LaTV-TSVD puede abortarse de forma aguda conmaniobras vagales, masaje del seno carotídeo, adenosina intravenosa (6-18 mg) o verapamilo intravenoso (5-10 mg). La eficacia de los fármacos antiarrítmicos (bloqueadores beta, antagonistas delcalcio) en pacientes con TV-TSVD es de alrededorde un 25-50%. Teniendo en cuenta la tasa de éxitosa largo plazo (> 90%) y su baja incidencia de complicaciones mayores (< 1%)93,97, la ablación por catéter se está convirtiendo en el tratamiento de primera línea en pacientes con TV-TSVD.
CARDIOPATÍAS CONGÉNITAS (TETRALOGÍA DE FALLOT)
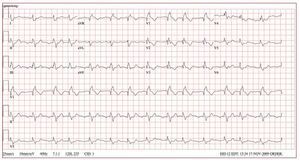
Pueden observarse alteraciones del ritmo en laevolución natural de las cardiopatías congénitas(CPC), así como tras la cirugía a corazón abierto104. Hay un número creciente de pacientes con CPC quellegan a una edad adulta, gracias al éxito de las intervenciones quirúrgicas correctoras105. Alrededorde un 40-50% de los adultos con CPC sufren algúntipo de arritmia. La incidencia elevada de arritmiascardiacas en los adultos con CPC puede deberse alos cambios de presión/volumen anormales y conmás frecuencia a circuitos de reentrada creados porparches del tabique y líneas de sutura105,106. Las taquicardias supraventriculares, como el aleteo auricular y/o la fibrilación auricular, son frecuentes enlas CPC (fig. 9).

Fig. 9. El electrocardiograma de 12 derivaciones muestra un aleteo auricular enuna mujer de 42 años a la que se habíapracticado una corrección de una tetralogía de Fallot 31 años antes. Esta mujerfue ingresada en nuestro centro a causade palpitaciones. Se observó aleteo auricular derecho de sentido antihorariocaracterístico (dependiente del istmo cavotricuspídeo) durante un estudio de electrofisiología, y se trató con ablación porradiofrecuencia. El electrocardiogramade 12 derivaciones tras la ablación continuaba mostrando un bloqueo de ramaderecha del haz de His y una desviacióndel eje a la derecha que reflejaban la disfunción del corazón derecho.
La TV se observa en un número de pacientesmenor, sobre todo en los que presentan una tetralogía de Fallot (TdF). Este trastorno incluye cuatrocaracterísticas: estenosis infundibular subpulmonar,comunicación interventricular, dextroposición de laaorta (aorta acabalgada) e hipertrofia ventricularderecha. La intervención quirúrgica incluye unaventriculotomía derecha y una reparación de la comunicación interventricular mediante parche. Estas lesiones hacen que haya un sustrato para laarritmia. La reparación de la comunicación inter-ventricular mediante un parche proporciona unobstáculo anatómico fijo alrededor del cual puedenproducirse arritmias de reentrada.
Tras la reparación de una TdF, la TV sostenidatiene una prevalencia de un 4-7%107 y suele consistiren una taquicardia de reentrada del TSVD con unamorfología de BRIHH. Las AV no sostenidas sedan en hasta un 60% de los registros Holter.
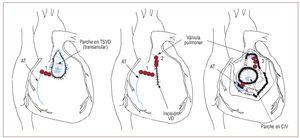
En estos pacientes hay un riesgo persistente deMSC tardía, que tiene una incidencia estimada deun 0,5-8,3%108,109. La mayor edad en el momento dela reparación inicial, la presencia de una insuficiencia pulmonar moderada o grave, los antecedentes de TV sostenida, la disfunción ventricularmoderada o grave y la duración del QRS ≥ 180 msson factores que predicen el riesgo de MSC109. La TV no sostenida en pacientes con TdF carece devalor predictivo respecto a TV sostenida ulterior ola MSC108. Los pacientes generalmente presentancomo manifestación inicial palpitaciones y conmenor frecuencia síncope o presíncope. Los fármacos antiarrítmicos como amiodarona y sotalolsuelen utilizarse en estos pacientes como tratamiento de primera línea. En los pacientes con unaTV sintomática refractaria a la medicación, puedeutilizarse ablación con radiofrecuencia. Se utilizanmapas convencionales y electroanatómicos paraidentificar el circuito de reentrada. La elaboraciónde un mapa electroanatómico puede ser crucialpara el éxito, sobre todo en las TV mal toleradas.Se ha demostrado que la existencia de un istmoentre un área de cicatriz/parche de la pared anteriordel TSVD y el anillo tricuspídeo (fig. 10) es la principal causa de las TV (75%) en los pacientes sintomáticos a los que se practica ablación con radiofrecuencia tras la reparación de la TdF. A largo plazopuede alcanzarse una tasa de éxito de alrededor del 90% de los pacientes110. En los pacientes con unaMSC abortada y TV en los que no puede aplicarsela ablación, está indicado un DAI.

Fig. 10. Localización de los límites anatómicos (línea azul) de la taquicardia ventricular derecha tras la reparación de un defecto cardiaco y los consiguientes istmos anatómicos (líneas rojas, números 1 a 4). AT: anillo tricuspídeo; CIV: comunicación interventricular; TSVD: tracto de salida ventricular derecho;VD: ventrículo derecho. Modificado con permiso de Zeppenfeld et al107.
VENTRÍCULO DERECHO, ANOMALÍASDEL ECG Y MUERTE SÚBITA CARDIACAEN DEPORTISTAS
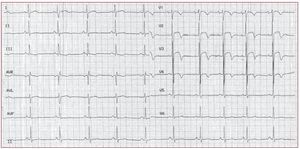
Se ha estimado que las AV en pacientes con enfermedades del VD, como el SB y la DAVD, suponenhasta un 10-30% de las MSC de adultos jóvenes dela población general. Este porcentaje es aún másalto en los deportistas jóvenes2. España se ha vistoafectada recientemente por la muerte súbita de unjoven futbolista nacional afectado por una DAVD.Se ha descrito que en España cada año fallecen20-35 deportistas jóvenes por muerte súbita. LaMSC de un individuo joven y por lo demás aparentemente sano es siempre un suceso trágico que puedeser incluso más cruel cuando sucede a la vista de millones de personas. Se ha demostrado que el examende detección sistemático previo a la participación enla actividad deportiva logra reducir la MSC en lapoblación de deportistas111. El ECG de doce derivaciones en individuos entrenados con frecuencia esanormal. Las anomalías frecuentes y relacionadascon el entrenamiento son la bradicardia sinusal, elbloqueo de primer grado y de Wenckebach, elBRDHH incompleto, la hipertrofia ventricular izquierda y el patrón de repolarización temprana.Estas anomalías fisiológicas reflejan un remodeladodel corazón y el cambio del balance del sistema autónomo en los individuos con un entrenamiento intenso y regular. Sin embargo, en un pequeño porcentaje de los casos112 (8%), las alteraciones del ECGpueden ser intensas y difusas. Son necesarios una interpretación cuidadosa y un estudio diagnóstico(fig. 11) en estos casos específicos, con objeto de descartar una enfermedad que ponga en peligro la vida, sobre todo en presencia de síncopes o antecedentesfamiliares de MSC. En las cardiopatías de causa genética (DAVD, SB, miocardiopatía hipertrófica,síndromes de QT largo-corto) existe un grado variable de influencia del ambiente, que puede agravaro incluso desenmascarar el fenotipo de la enfermedad. Así, en pacientes con DAVD, el ejercicioagrava las anomalías de los contactos intercelularesdeterminadas genéticamente113,114. El ejercicio, sobretodo durante la fase de recuperación, puede hacertambién que se manifieste un ECG de SB de tipo 1y, por consiguiente, puede exponer al paciente a unasituación que ponga en peligro su vida81,115-117. En general, a los pacientes con DAVD y SB se les debe aconsejar que eviten el ejercicio extenuante y el entrenamiento enérgico, y se los debe excluir de la participación en deportes de competición o profesionales.

Fig. 11. Electrocardiograma de un futbolista de competición de 17 años asintomático, en reposo y sin signos de hipertrofiaventricular izquierda u otras anomalíasestructurales en la ecocardiografía, quemuestra anomalías de la repolarizaciónnotables, incluida una elevación del segmento ST y una inversión de la onda Ten las derivaciones precordiales V1-V4. Ante este patrón, es obligado descartartrastornos cardiacos con peligro para lavida, como la miocardiopatía hipertrófica,la displasia arritmogénica ventricular derecha, la miocardiopatía dilatada, la estenosis valvular aórtica y las canalopatías(síndrome de Brugada, síndrome de QTlargo y corto).
La implantación de un DAI se debe valorar seriamente tras una estratificación cuidadosa del riesgo.Hay controversia respecto a si los deportistas a losque se ha colocado un DAI pueden o no continuarpracticando los deportes de competición. Según laguía de Bethesda118 los pacientes portadores de unDAI pueden participar tan sólo en actividades de«clase IA», como los bolos o el golf. En cambio,otros datos más recientes indican que, aunque lasdescargas durante la práctica deportiva se describencon frecuencia, los acontecimientos adversos importantes son muy poco frecuentes119. Así pues, lasrecomendaciones médicas respecto a la práctica deportiva continúan siendo aún muy diversas. Hay varias razones teóricas y prácticas para limitar la práctica deportiva a los individuos portadores de un DAI,pero de hecho no hay datos que demuestren que losdeportes son peligrosos para todos los pacientes conDAI, y es probable que la decisión de cada deportistadeba basarse en una elección informada.
ABREVIATURAS
AV: arritmias ventriculares.
CPC: cardiopatías congénitas.
DAVD: miocardiopatía/displasia arritmogénicade ventrículo derecho.
MSC: muerte súbita cardiaca.
SB: síndrome de Brugada.
TSVD: tracto de salida ventricular derecho.
TV-TSVD: taquicardia ventricular de tracto desalida ventricular derecho.
VD: ventrículo derecho.
Full English text available from: www.revespcardiol.org
Correspondencia: Dr. L. Capulzini.
Heart Rhythm Management Center. UZ Brussel-VUB.Laarbeeklaan 101. 1090 Brussels. Bélgica.
Correo electrónico: luciomassimo@hotmail.com