Palabras clave
INTRODUCCION
Es bien sabido ya desde los años sesenta que el infarto de miocardio, y en general toda la patología aterosclerosa, tiene un componente hereditario importante. Así se desprende de los numerosos estudios realizados en familias y en gemelos1-3, que han descrito el riesgo que comporta tener un hermano gemelo o un pariente afectado de enfermedad coronaria (tabla 1).

La identificación de los genes responsables del aumento del riesgo, sin embargo, ha sido un proceso lento y difícil, acelerado en la última década gracias a los avances de la biotecnología, que han facilitado la detección de los cambios en la secuencia del ADN que pueden tener un efecto patógeno3. Estos cambios, que llamamos mutaciones o polimorfismos, pueden ser muy sutiles: unas veces se trata de la sustitución de un simple nucleótido (un simple aminoácido en la proteína codificada) entre miles (SNP o single nucleotide polimorphism); en otras se produce la inserción o deleción de un segmento, o la repetición de unas secuencias en tándem (VNTR, número variable de tandem repeats). Puede ocurrir en el exón o segmento codificante, en el intrón o en la zona del promotor del gen.
Mutaciones y polimorfismos son en realidad palabras sinónimas. Ambos se caracterizan por la coexistencia de dos variedades o alelos del mismo gen, el alelo natural o silvestre (wild type) y el alelo mutante. Pero suele reservarse el término de mutaciones para los cambios que alteran gravemente la función de la proteína o enzima codificada y basta un gen para provocar una enfermedad (enfermedad monogénica o mendeliana). Son raras y siguen las leyes de la herencia mendeliana, dominante o recesiva. En cambio, se denominan polimorfismos si las variaciones son comunes (por definición ocurren en más del 1% de la población) y la afectación funcional es modesta o mínima, pero supone una minusvalía (un factor de riesgo genético) cuando el organismo debe enfrentarse con un mayor esfuerzo metabólico o un problema ambiental, como puede ser una dieta rica en colesterol, el estrés o el tabaco (factor de riesgo ambiental). Muchos polimorfismos, sin embargo, no tienen consecuencia funcional alguna. La suma de varios polimorfismos desfavorables puede facilitar la aparición de una enfermedad (en este caso poligénica), cuya manifestación requiere a menudo la presencia de un marco ambiental propicio (enfermedad multifactorial).
La arteriosclerosis es su ejemplo más clásico (fig. 1) y se han descrito un buen número de polimorfismos que pueden estar implicados en su patogenia (tablas 2 y 3)4-6. El método que se utiliza para describir un alelo de riesgo consiste en identificar un gen polimórfico como posible candidato y demostrar su asociación estadística con el fenotipo clínico (enfermedad coronaria) y con el fenotipo intermedio (valores biológicos que dependen de la enzima).

Fig. 1. Etiología de la aterosclerosis coronaria: factores genéticos y ambientales. En cursiva y dentro de la figura elíptica, se indican los genes polimorfos. Las flechas finas señalan las interacciones entre factores ambientales y genéticos. AGN: gen del angiotensinógeno; apo-E: apolipoproteína E; AT1: receptor de la angiotensina; ECA: angina conversiva de la angiotensina; FGN: fibrinógeno; HDL: lipoproteína de alta densidad; HTA: hipertensión arterial; IRS-1: sustrato del receptor de la insulina; LDL: lipoproteína de baja densidad; Lp(a): lipoproteína (a); MTHR: tetrahidrofolatorreductasa; NOS: sintasa del óxido nítrico; NADH: NADH oxidasa; PAI-1: inhibidor del activador del plasminógeno.


GENES RELACIONADOS CON LAS LIPOPROTEINAS DE BAJA DENSIDAD
La lista empieza con las variaciones genéticas que afectan la concentración plasmática de las lipoproteínas de baja densidad, ricas en colesterol (LDL), cuyo depósito y efecto lesivo sobre la pared vascular constituye el fenómeno central de la aterogenia4,5 (fig. 2).

Fig. 2. Genes del metabolismo de las lipoproteínas de baja densidad (cLDL). Los genes implicados en la etiología de la aterosclerosis coronaria se indican en cursiva dentro de un recuadro. En el encarte: esquema de una partícula esférica de LDL, el agente específico del ateroma, con su molécula de apo B filamentosa que la rodea, a la que se añade una cadena con anillos kringle IV, la apo(a), para formar el complejo Lp(a). Flecha 1: vía de formación y transporte de las LDL a los receptores LDL-R de los hepatocitos o las células periféricas; flechas 2: transporte de las LDL, LDL pequeñas (tipo B) y Lp(a) a la pared vascular, donde, tras sufrir oxidación, son captadas por los receptores SR-B1 de los macrófagos. Apo-B, E, (a): apolipoproteínas B, E y (a); IDL: lipoproteínas de densidad intermedia; LDL: lipoproteínas de baja densidad; LDL-R: receptores de las LDL; SR-A: receptores basureros (scavenger receptors); VLDL: lipoproteínas de muy baja densidad.
Receptor de las LDL
El primer gen que se relacionó con el infarto de miocardio fue el gen que codifica los receptores de las LDL (R-LDL)7. Las mutaciones de este gen impiden totalmente la síntesis del receptor en los hepatocitos («mutación nula») o lo deforman de tal manera que son incapaces de captar las LDL circulantes para su eliminación por la bilis. En consecuencia, se produce la acumulación de estas partículas, que caracteriza la hipercolesterolemia familiar (HF). En su forma heterozigota, relativamente frecuente (1 de cada 500 individuos), la cifra del colesterol aumenta hasta 300-500 mg/dl, y esta cifra se duplica en los pacientes homozigotos, que son excepcionales (1/1.000.000). Esta concentración tan elevada facilita la entrada pasiva de las macromoléculas a través de las uniones de las células endoteliales al espacio subendotelial, donde sufren las modificaciones químicas (peroxidación lipídica, glucación) que las convierten en partículas proinflamatorias, procitotóxicas y aterogénicas4-7.
Poco después de descubrir el defecto molecular de la hipercolesterolemia familiar, que les valió el Premio Nobel en 1984, Goldstein y Brown describieron las primeras mutaciones causales de la enfermedad8. Hoy se conocen más de 230 (véase http://www.ucl.ac.uk/fh). Estas mutaciones presentan una gran variabilidad regional. En nuestro país, en la zona mediterránea, el 55% de las mutaciones detectadas en pacientes heterozigotos tenía una «mutación nula». El resto tenían mutaciones de un simple nucleótido, siendo las más frecuentes las 1146 G/A, 1301 C/G, y 829 G/A9.
La gravedad de la hipercolesterolemia y de la enfermedad coronaria también es muy variable: unas veces aparece el infarto de miocardio en edades muy jóvenes; en cambio, algunos pacientes heterozigotos alcanzan la séptima u octava década de la vida sin complicaciones. Ello depende: a) del tipo de la mutación (las «mutaciones nulas» son las más graves)9,10; b) de la interacción con otros factores genéticos, el más importante de los cuales parece ser una cifra favorable de lipoproteínas de alta densidad (HDL) plasmática, debida quizás a la herencia adicional de una lipoproteinlipasa mutante heterozigota9, o c) de la importancia de los factores ambientales (el contenido de colesterol y grasas de la dieta), como indica el distinto pronóstico de los familiares que han emigrado cuando se compara con el de los autóctonos4.
Se conoce también una variante común, el polimorfismo Pvu II en el intrón 15 (que permite el corte por la enzima Pvu II y da lugar a fragmentos de distinta longitud en el estudio electroforético: polimorfismo RFLP (restriction fragment lengh polymorphism). Poco se sabe de esta variante, pero al parecer influye en la concentración de LDL y en el riesgo coronario11.
Apolipoproteína B (apo-B100)
Otra causa de hipercolesterolemia familiar, menos grave, es la mutación Arg3500Gln (y excepcionalmente la Arg3531Cys) de la apolipoproteína B (apo-B100), la molécula que actúa de ligando para los receptores LDL (defecto familiar de la apo-B o FDB). La mutación impide su reconocimiento por el receptor LDL y dificulta su eliminación del plasma. Su prevalencia es de un caso cada 250-1.000 individuos4,6,7.
En cambio, se conocen algunos polimorfismos comunes del gen de la apo-B cuya asociación con la hipercolesterolemia y la enfermedad coronaria es muy sugestiva, pero los resultados contradictorios de algunos estudios la ponen en duda. Se trata de los polimorfismos Sp I/D (inserción/deleción en el péptido señal de la apo-B), EcoRI y MspI (polimorfismos RFLP)12,13.
Algunas mutaciones raras (0,3/1000) codifican formas truncadas de apo-B100 (apo-B55) y dan lugar a cifras bajas de colesterol (Hipobetalipoproteinemia o HBLP), que tienen probablemente un efecto protector contra la enfermedad coronaria4,7.
Polimorfismos de la apolipoproteína E (apo-E)
La apo-E es una proteína que comparte con la apo-B la función de ligando de las lipoproteínas ricas en triglicéridos (lipoproteínas de muy baja densidad [VLDL] y de densidad intermedia [IDL]). Se presenta en tres versiones principales: la apo-E3, la isoforma natural, la más comun, y dos formas mutantes, la apo-E2, en la que se ha sustituido arginina por cisteína en la posición 158, y la apo-E4, que cambia cisteína por arginina en posición 112.
El alelo polimórfico apo-E4 tiene una menor afinidad que la apo-E3 para los receptores apo-B/E y se asocia a un ligero aumento de colesterol y los triglicéridos plasmáticos. Su relación con la enfermedad coronaria es de las más firmes14-16. Los portadores de este alelo tienen un riesgo coronario de un 40% superior al de los alelos apo-E3 o apo-E215. El estudio 4S (Scandinavian Simvastatin Survival Study) ha podido comprobar el efecto letal del alelo apo-E4 en pacientes que han padecido un infarto de miocardio, pues la mortalidad asociada a este alelo fue del 15,7% a los 5,4 años de seguimiento, cuando en el resto de pacientes fue del 9%. Además, el alelo apo-E4 permitía predecir una buena respuesta al tratamiento hipolipemiante: la mortalidad de los pacientes portadores de este alelo se redujo del 15,7 al 6%, en tanto que en el resto la reducción de la mortalidad fue sensiblemente menor (del 6 al 5,1%)16. Este dato confirma que la respuesta al tratamiento con estatinas depende del genotipo apo-E, como se había descrito previamente17. Se ha señalado también que el alelo apo-E4 disminuye la probabilidad de alcanzar edades muy avanzadas, pues la prevalencia en nonagenarios (11,6%) es inferior a la de los individuos de mediana edad (22%)18. Por otra parte, el alelo apo-E4 se acompaña de un ligero aumento del riesgo de padecer la enfermedad de Alzheimer4,7.
En cambio el alelo apo-E2 parece tener un efecto favorable. Un reciente estudio comparativo sobre el efecto de la ingesta rica en colesterol en pacientes con distintos genotipos apo-E1, B, C-III, E y R-LDL ha demostrado que los individuos con el alelo apo-E2 tienen los niveles más bajos de cLDL y no responden al aumento del colesterol en la dieta, mientras que las mutaciones de la apo B tienen las elevaciones más altas, sobre todo si coexisten con el alelo apo-E419. El alelo apo-E2 se ha relacionado también con: a) la hipobetalipoproteinemia (cLDL < 70 mg/dl), cuya prevalencia en el Framingham Off-Spring Study fue del 2%. La otra causa de hipobetalipoproteinemia, la apo B truncada, es mucho menos frecuente13, y b) la disbetalipoproteinemia familiar (hiperlipoproteinemia tipo III), una enfermedad rara que afecta a pacientes con el alelo apo-E2, está producida por una deficiencia completa de la apo-E y se asocia sobre todo con enfermedad vascular periférica.
Polimorfismo de la apo(a)
Las lipoproteínas (a) -Lp(a)- son un pequeño subgrupo de partículas LDL que contienen, además de una molécula de apo-B100, una molécula de apo(a), que es extraordinariamente parecida al plasminógeno, pero carece de su efecto mediador de la fibrinólisis. Esta molécula presenta numerosas secuencias repetidas (hasta un máximo de 37) del anillo kringle IV (kringle en recuerdo de una pasta danesa), el mismo que se repite 5 veces en el plasminógeno4,6,20.
Las Lp(a) son particularmente nocivas, porque: a) son atraídas por la fibrina depositada en las lesiones de la íntima; b) tienen un efecto antifibrinolítico, pues compiten con el plasminógeno y bloquean su efecto potenciador de la lisis del coágulo en los accidentes coronarios agudos, y c) anulan el efecto activador de la plasmina sobre el factor TGF-β que tiene una importante efecto inhibidor de la proliferación y migración de las células musculares lisas7,20.
La concentración plasmática varía según los individuos entre menos de 1 y más de 100 mg/dl (10 mg/dl de promedio) y guarda una estrecha relación inversa con el número de anillos de la apo(a). La cifra, en cambio, es muy constante en cada individuo y poco susceptible de modificación dietética o farmacológica, ya que el factor genético es responsable del 90% d e la variabilidad7,20.
Como han confirmado ya numerosos estudios epidemiológicos, la presencia de concentraciones elevadas de Lp(a) (> 30 mg/dl) se asocia a un aumento de la incidencia de infartos de miocardio, de reestenosis y de accidentes vasculares cerebrales4,21,22. Se ha señalado incluso que puede ser la causa principal del 25% de los infartos de miocardio en EE.UU.22. Tiene además un significado pronóstico desfavorable. En los pacientes con infarto de miocardio del ensayo clínico escandinavo 4S la mortalidad a los 5,4 años de los pacientes con una concentración elevada de Lp(a) era el doble que en el resto. Más llamativo aún, si coincidían dos polimorfismos desfavorables, el de la Lp(a) y el alelo polimórfico apo-E4, la mortalidad era más del triple, lo que es un buen ejemplo del efecto amplificador de la interacción entre dos genes. Por otra parte, la presencia del polimorfismo apo(a) predecía una respuesta más eficaz al tratamiento con simvastatina, con una reducción de la mortalidad del 50%. Esta reducción ascendía al 80% si el paciente poseía ambos polimorfismos desfavorables, el de la apo(a) y el de la apo-E16.
GENES RELACIONADOS CON LAS LIPOPROTEINAS DE ALTA DENSIDAD (HDL) Y EL TRANSPORTE INVERSO
El estudio Framinghamya ya dejó que la baja concentración plasmática de las HDL es uno de los factores de riesgo coronario más potentes, tanto o más que las cifras altas de LDL. Ello se atribuye al importante papel protector de estas partículas, que son responsables del transporte inverso del colesterol desde la pared vascular a los receptores hepáticos SR-B1 (scavenger receptor tipo B1) para su eliminación por la bilis23. Las concentraciones de HDL plasmáticas y la eficacia del transporte inverso dependen de la actividad física o el consumo de alcohol, pero tienen componente genético importante que se cifra en un 35-50%, en el que están implicados buen número de polimorfismos4,7 (fig. 3).

Fig. 3. Genes del metabolismo de las HDL y los triglicéridos (TG). En cursiva se indican los genes polimórficos que codifican las enzimas y los receptores del metabolismo de los TG y las HDL, y se han implicado en la aterogénesis. Flecha 1: vía del transporte inverso del colesterol desde la placa de ateroma al hígado, para su eliminación por la bilis. Su eficacia depende de las producción de las enzimas que facilitan la extracción del colesterol de los macrófagos (ABC1) y su incorporación a las partículas de HDL (LCA, HL), de los componentes de las HDL (apoproteínas A1-CIII-A4, PONA), o de los receptores hepáticos específicos (SR-B1); flecha 2: vía de transporte alternativo del colesterol de las HDL a las VLDL e IDL, regulada por la enzima CETP. En el recuadro: vía de transformación de las partículas ricas en triglicéridos, VLDL y IDL, en LDL, bajo la acción de la lipasa lipoproteica (LPL). ABC1: transportador ABC1; apo: apolipoproteína; CETP: proteína de transferencia del colesterol esterificado; HL: heparinlipasa; LCAT: enzima lecitín aciltransferasa; IDL: lipoproteínas de densidad intermedia; LDL: lipoproteínas de baja densidad; LDL-R: receptores de las LDL; SR-B1: receptores basureros (scavenger receptors); VLDL: lipoproteínas de muy baja densidad.
Lipasa lipoproteica y triglicéridos
La lipoproteinlipasa (LPL) es la principal enzima del catabolismo de las lipoproteínas ricas en triglicéridos (VLDL e IDL). Tiene dos funciones principales: a) cataliza la liberación de los triglicéridos (TG) de las VLDL cuando pasan por los capilares y las degrada a remanentes o IDL, y luego a LDL, y b) una vez realizada su función enzimática, se desprende de su anclaje en el endotelio y actúa de ligando de las IDL para su captación y eliminación por los receptores hepáticos4,7,23.
El gen de la lipoproteinlipasa es particularmente propenso a las mutaciones. Se han descrito cerca de 40 (véase: www.ncbi.nlm.nih.gov/omim). Unas se localizan en el segmento terminal C (residuos 313 a 448) y alteran la función de ligando, y otras en el dominio N (del residuo 1 al 312), deprimen la función enzimática y disminuyen el catabolismo de VLDL y las IDL, lo que ocasiona la acumulación de estas partículas en el plasma (el aumento de los TG) y la disminución de las HDL4,7.
Se conocen tres mutaciones localizadas en el dominio N. La Asp9Asn y la Asn291Ser, cuya prevalencia en su forma heterozigota es del 3-5%, se asocian a un aumento del 20-30% en los TG en el plasma, una reducción del cHDL ~‾ 10 mmol/l y un aumento del riesgo de cardiopatía isquémica (riesgo relativo ~‾ 1,3). La Gly188Glu, la menos frecuente (1/1.000), es la que más deprime la actividad enzimática, induce un mayor aumento de los TG (80%), una mayor disminución del cHDL (25 mmol/l) y un aumento más importante del riesgo coronario (riesgo relativo de 5/1)24.
El mecanismo de la relación entre el aumento de los TG del plasma (el aumento de las VLDL y IDL) y la enfermedad coronaria sigue siendo un tema controvertido. Se han propuesto varias explicaciones: a) se sospecha que la incorporación a la pared arterial de las moléculas más pequeñas de VLDL y de IDL (remanentes de VLDL), que también contienen colesterol aunque en menor proporción, tienen un efecto aterogénico comparable al de las LDL25. Pero es difícil disociar este efecto de las importantes modificaciones metabólicas que inducen los TG (las VLDL e IDL) en otras lipoproteínas, como las HDL y las LDL; b) la disminución de las HDL, que guarda una estrecha relación inversa con los TG, atribuible al efecto de los TG (y la insulina) sobre el catabolismo de las apo-A1, la apolipoproteína propia de las HDL; o a la mayor actividad de la enzima CETP, que desvía el transporte del colesterol a otras vías alternativas en detrimento del transporte inverso de la HDL (véase más adelante), o c) el aumento de las LDL pequeñas y densas (LDL tipo B), que tienen un gran potencial aterogénico debido posiblemente a su mayor capacidad de penetración en la pared vascular y su fácil oxidación25-27.
Los análisis de regresión múltiple parecen confirmar que los TG (las VLDL y IDL) son un tercer factor de riesgo epidemiológico independiente de las LDL y las HDL, aunque los resultados no son muy llamativos. El efecto es más aparente cuando se determinan los TG posprandiales, que reflejan mejor la capacidad de respuesta metabólica del individuo27. Un reciente metaanálisis de 17 estudios resume los datos que apoyan este concepto24.
Estos polimorfirmos de la lipoproteinlipasa presentan una interesante interacción con la obesidad, como ha apuntado el estudio EARS, pues la elevación de los TG inducida por el exceso de peso se acentúa en
los portadores de la mutación LPL-291S26.
Una cuarta variante, el polimorfismo exónico Ser447Ter, que se localiza en el terminal C y afecta la función de ligando, reviste un interés especial porque, aparte de ser la más frecuente (su prevalencia en la población general es del 20%), se asocia a una concentración ligeramente más elevada de las HDL (0,04 mmol/l), una reducción de los TG (8%) y un menor riesgo de enfermedad coronaria (riesgo relativo: 0,8). Este efecto protector se atribuye a su mayor afinidad por los receptores, que facilita la eliminación de las partículas remanentes de VLDL24. Esta variación está íntimamente ligada al polimorfismo LPL/HindIII (polimorfismo RFLP)4,7.
Proteína de transferencia de los ésteres de colesterol (CETP) y alcohol
Las HDL poseen dos enzimas esenciales para el transporte inverso: la LCAT (lecitina-colesterol aciltransferasa), que facilita la captación del colesterol de la pared vascular y da comienzo a la vía del transporte inverso, y la CETP (cholesterol ester transfer protein), que promueve el trasiego del colesterol de las HDL y LDL (ricas en colesterol) a las VLDL e IDL (ricas en TG), e inicia una vía catabólica alternativa para su eliminación por el hígado, menos eficiente que la del transporte inverso4,7.
Las variaciones conocidas de la LCAT no parece que tengan mayor trascendencia, aunque se ha descrito una mutación, LCAT-Gly230Arg, responsable del 5% de los casos de HDL muy bajas28. Se conoce en cambio un polimorfismo TaqI-B del intrón 1 del gen de la CETP (CETP/Taq1), que aumenta la actividad de la enzima y tiene un efecto proaterogénico29. Este efecto se atribuye a que: a) la activación del CETP desvía el colesterol del transporte inverso a la vía alternativa, con lo que disminuye las HDL; b) enriquece el colesterol de las partículas que transportan TG (VLDL y IDL), lo que aumenta su potencial aterogénico, y c) empobrece de colesterol las LDL, y las convierte en partículas más pequeñas y densas (LDL patrón B), especialmente aterogénicas4,7,26. Se sospecha que la elevación de los TG y la hipertrigliceridemia posprandial (que dura 8 h y, por tanto, estamos casi siempre en período posprandial a este respecto) desempeñan un papel importante en la génesis de la aterosclerosis, en parte debido a su capacidad de activar la CETP28.
Como ha demostrado el estudio REGRESS (Regression Growth Evaluation Statin Study), los portadores del genotipo B1B1 (frecuencia del 35%) tienen una actividad elevada de la enzima, cifras altas de TG y bajas de HDL, y presentan una mayor progresión de la aterosclerosis coronaria que los portadores de B1B2 y B2B2. La presencia de este alelo permite además predecir una mejor respuesta de las lesiones angiográficas al tratamiento con estatinas29.
Este polimorfismo presenta una interacción muy notable con el alcohol. Como ha señalado el estudio ECTIM (Etude Control-Temoigne l'Infarctus de Myocarde), la relación entre el polimorfismo CETP y las HDL sólo se manifiesta en los individuos que beben. En cambio, no se observa en los abstemios. Esto indica que el efecto beneficioso de la ingesta moderada de alcohol sobre las HDL podría estar relacionado con su efecto depresor sobre la CETP30.
Paraoxonasa (HDL-PON1)
La paraoxonasa/arylesterasa es una enzima específica de las HDL (asociada a las apo-A1) capaz de hidrolizar los peróxidos lipídicos y destruir las moléculas proinflamatorias producidas por la oxidación de las LDL, por lo que se sospecha que puede desempeñar un papel muy relevante en la etiopatogenia de la aterosclerosis31. Se detecta normalmente en la pared arterial y su concentración aumenta de forma masiva en las placas de ateroma, posiblemente en respuesta al aumento del estrés oxidativo31.
La actividad enzimática de la PON1 presenta una variación interindividual de un 10-40%, atribuible en parte a la presencia de dos polimorfismos: el PON1-192, con sustitución de arginina por glutamina en el codón 192 (A/G 192), que da lugar a una isoenzima R con arginina y una isoenzima Q mutante, y el PON1-55 que sustituye leucina (L) por metionina (M) en el codón 55. Se ha comprobado mediante experimentos de coincubación que la capacidad de las partículas HDL para proteger contra la modificación oxidativa de las LDL es claramente superior en las partículas procedentes de homozigotos QQ/MM que las procedentes de homozigotos RR/LL32.
A pesar de que algunos estudios como el ECTIM33 niegan la asociación del polimorfismo 192 con la enfermedad coronaria, otros más recientes señalan que el genotipo RR/LL puede ser un importante factor de riesgo independiente, sobre todo en pacientes con diabetes tipo 2, en los que la actividad enzimática y los niveles plasmáticos de PON1 están habitualmente disminuidos34. El estudio REGICOR ha confirmado que el alelo R aumenta el riesgo de infarto en pacientes diabéticos35. Cabe la posibilidad de que el aumento de riesgo (que puede ser del 60%) sólo se detecte en los fumadores36. Esta interacción gen/ambiente puede atribuirse a que el tabaco tiene una acción deletérea sobre la paraoxonasa, que anula el efecto de las diferencias interindividuales.
Transportador ABC1
El transportador ABC1 es una proteína que interviene de manera decisiva en la salida del colesterol de las células, pues forma un canal que permite su paso al exterior de la membrana, donde se transfiere a las partículas que nacen de HDL, previa esterificación por la LCAT. La confirmación de que la enfermedad de Tangier, una enfermedad mendelinana rara que cursa con niveles muy bajos de HDL y enfermedad coronaria prematura, se debe a una mutación del gen del transportador ABCA1 propició la búsqueda de polimorfismos comunes de este gen que pudieran influir en el riesgo coronario4. Se ha identificado recientemente un polimorfismo -477T/C ABCA1, cuyos genotipos TT y TC se asocian a una reducción modesta de las cifras de HDL y apo-A1, pero muestran una correlación muy fuerte con la gravedad de la enfermedad coronaria a juzgar por el número de lesiones en la angiografía37.
Polimorfismos de la apoproteínas AI/CIII/AIV y la lipasa hepática
Se han identificado variantes de la lipasa hepática y en los loci apo-A1/CIII/AIV (cuyos genes codifican componentes estructurales de las HDL), que pueden influir en la concentración de las HDL y podrían modificar la frecuencia de la enfermedad coronaria38. Cabe recordar que existe una enfermedad mendeliana rara, la deficiencia de apo-AI, que en su forma homozigota comporta una ausencia virtual de HDL (alelo nulo) y da lugar a una enfermedad coronaria prematura grave4,7.
HIPERTENSION ARTERIAL. SISTEMAS RENINA-ANGIOTENSINA-ALDOSTERONA Y SIMPATICOADRENÉRGICO
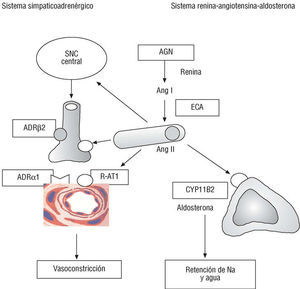
La hipertensión arterial y la hipertrofia ventricular se encuentran entre los primeros factores de riesgo coronario descritos en el estudio Framingham. Es pues lógico que se hayan sometido a un intenso escrutinio las variantes moleculares de los genes de los sistemas renina-angiotensina-aldosterona y simpaticoadrenérgico (fig. 4).

Fig. 4. Genes de los ejes simpaticoadrenérgico y renina-angiotensina-aldosterona. En cursiva y en recuadro se indican los genes polimórfios que codifican enzimas o receptores implicados en la patogenia de la enfermedad coronaria. AGN: angiotensinógeno; Ang: angiotensina; ADR: receptor adrenérgico; CYP11B2: enzima sintasa de la aldosterona; ECA: enzima conversiva de la angiotensina; R-AT1: receptor AT1 de la angiontensina II; SNC: sistema nervioso central.
Angiotensinógeno
El polimorfismo M235T del gen del angiotensinógeno (AGT) es el que presenta una relación más clara con la hipertensión, aunque el efecto parece ser débil. Su relación con la enfermedad coronaria, en cambio, es dudosa39-41. Se ha señalado que el genotipo 235 TT duplica el riesgo de cardiopatía coronaria y de infarto de miocardio40, aunque el Copenhagen City Heart Study, con más de 2.800 pacientes, ha sido negativo41.
Polimorfismo I/D de la enzima conversiva de la angiotensina (ECA)
Una de las variantes genéticas mejor estudiadas es el polimorfismo inserción/deleción (I/D) del gen de la ECA, la ectoenzima de las células endoteliales que interviene en la generación de angiotensina II y la degradación de la bradicinina, dos péptidos vasoactivos con importantes efectos sobre la proliferación celular y la trombosis. Este polimorfismo se caracteriza por la presencia o ausencia de una secuencia Alu de 287 pares de bases en el intrón 16. Da lugar a tres genotipos distintos: II/ID/DD. Los portadores del alelo D presentan un aumento de la actividad de la ECA plasmática y la ECA cardíaca y tisular (que representa el 80% de la actividad de la ECA) y están expuestos, por tanto, a mayores niveles de angiotensina II. En el genotipo DD, presente en el 28-31% de los individuos (tabla 2), los niveles son el doble que en el genotipo II4,42.
Desde 1992, cuando se describió la asociación del genotipo DD con el aumento del riesgo de infarto de micoardio42, se han publicado numerosos estudios sobre la implicación del alelo D en la patogenia de la enfermedad coronaria y el infarto de miocardio43-48, el remodelado postinfarto49, la dispersión del QT postinfarto y la muerte súbita48,50, la restenosis postintervención percutánea51, la resistencia a la insulina y la diabetes52,53, la hipertensión arterial54 o la hipertrofia del ventrículo izquierdo55. El estudio ECTIM, basado en el registro MONICA, comprobó que el genotipo DD es un poderoso factor de riesgo de infarto de miocardio en pacientes menores de 65 años42, sobre todo en ausencia de otros factores de riesgo, en cuyo caso el riesgo se triplica. Se ha señalado que el genotipo DD puede ser el factor de riesgo coronario más importante en pacientes con diabetes tipo 253. En cambio, no parece que el gen de la ECA influya sensiblemente sobre la hipertensión arterial, por lo que cabe atribuir el efecto aterogénico de la angiotensina II a su acción metabólica (mitógena) sobre la pared vascular, más que a su efecto sistémico hipertensivo.
Por el contrario, algunos estudios de gran envergadura como el Physician's Health Study (> 3.500 médicos)43, o el realizado en 11.000 sujetos del estudio ISIS-346, niegan cualquier relación de este polimorfismo con el infarto de miocardio. De los dos grandes metaanálisis publicados, uno concluye positivamente44, y el otro, más reciente, que comprende a más de 32.000 individuos de raza blanca45, encuentra un aumento significativo del riesgo en los portadores del genotipo homozigoto DD. De manera que 9 años después de la descripción inicial42, y a pesar de que las pruebas experimentales y clínicas son muy convincentes a favor del papel causal de la ECA en la enfermedad coronaria, la implicación del polimorfismo ECA sigue en cuarentena.
Receptor tipo I de la angiotensina II
El polimorfismo A1166C del gen AT1R (del receptor tipo 1 de la angiotensina II) se asocia probablemente a un aumento de la sensibilidad del receptor AT1, a través del que se ejercen la mayoría de los efectos de la angiontensina II. El alelo C es más frecuente en pacientes hipertensos, y su relación con la susceptibilidad a la cardiopatía isquémica es indicativa47,48,56,57. La asociación del genotipo AT1R-CC con el ECA DD (el 2% de nuestra población) puede tener un efecto de tipo sinérgico47,48.
Quimasa (CMA)
La quimasa es una enzima de la pared vascular y del corazón que tiene la capacidad de convertir la Ang I en II y la big ET-1 en ET-1. Esta enzima permite explicar por qué la producción de Ang II tisular puede mantenerse normal a pesar de la inhibición completa de la ECA. Sin embargo, de momento no se ha podido confirmar la asociación del polimorfismo CMA A-1905G y el infarto de miocardio58,59.
Sintasa de la aldosterona (CYP11B2)
La aldosterona tiene importantes efectos cardiovasculares. Aparte de su efecto sobre la presión arterial y la volemia, es conocido su efecto sobre la fibrosis de la hipertrofia y del postinfarto60. No es de extrañar, pues, que haya despertado gran interés el estudio del polimorfismo C/T -344 de la región promotora del gen de la sintasa de la aldosterona (CYP11B2/ T344C), que se ha implicado en el remodelado ventricular de la hipertensión arterial61,62, o en el remodelado postinfarto, que no se ha confirmado63. En un reciente estudio preliminar de 16 polimorfismos genéticos, el análisis de regresión seleccionó el polimorfismo CYP11B2 como uno de los 9 polimorfismos relacionados con la enfermedad coronaria angiográfica64.
Otras variantes genéticas
Otros polimorfismos en estudio son los del gen del receptor adrenérgico β2 (ADRB2-Arg16Gly y Gln27Glu), de la α-aducina (Gly460Trp), una proteína de la capa interna de la membrana plasmática que puede ser responsable en parte de la hipertensión sensible a la sal; o de la endotelina (EDN1) y sus receptores A y B (EDNRA y EDNRB). Su consideración como genes de riesgo parece prematura5,65.
GENES RELACIONADOS CON LA RESISTENCIA A LA INSULINA (EL SINDROME METABOLICO)
El síndrome metabólico, también llamado síndrome de la resistencia a la insulina (o síndrome de Reaven), es un síndrome particularmente aterogénico, que intenta explicar la frecuente asociación de 4 factores de riesgo coronario clásicos (el «cuarteto de la muerte» de Kaplan)66,67:
1. Obesidad de predominio visceral (relación cintura/cadera > 1,02 cm), relacionada con la ingesta y el estrés, que podrían ser los factores ambientales necesarios para que se manifieste el síndrome.
2. Hipertrigliceridemia, disminución de las HDL y aumento de las LDL tipo B (la llamada «tríada lipídica» o dislipemia aterogénica). El aumento de los TG (de hasta un 60%) se atribuye a un exceso de la producción hepática de las VLDL e IDL, estimulada por la mayor disponibilidad de los ácidos grasos libres del tejido adiposo. Pero cabría también atribuirla a una modificación de la actividad de la lipoproteinlipasa o la CETP (véase más arriba)68-70.
3. Resistencia a la insulina, hiperinsulinemia, intolerancia a la glucosa y diabetes tipo 2. La mayor disponibilidad de los ácidos grasos libres acentúa su consumo en el músculo esquelético en detrimento de la glucosa, cuyo aumento en la sangre estimularía la secreción, y pondría en evidencia la resistencia a la insulina. Entre los genes candidatos de la resistencia a la insulina se han señalado las mutaciones del receptor de la insulina (se han descrito más de 50), que serían causas monogénicas raras; o los polimorfismos de la FAB P2 (fatty acid binding protein, proteína de unión con los ácidos grasos), de la lipoproteinlipasa, del factor de necrosis tumoral alfa (TNF-α), o el genotipo DD de la ECA, pero sobre todo del gen IRS (insuline receptor substrate)71. La mayoría de los componentes del síndrome son premonitorios de la diabetes tipo 2, cuya incidencia alcanza el 15%, frente al 6,5% en la población general. La resistencia a la insulina puede preceder a la diabetes tipo 2 durante más de una década72.
La hiperglucemia es aterogénica porque facilita la glucación de las LDL y las convierte en partículas altamente lesivas, y porque promueve la aparición de productos finales de glucación avanzada (AGE), solubles, que interaccionan con receptores específicos del endotelio y aumentan la producción de radicales libres de oxígeno73.
4. Hipertensión arterial, relacionada con el aumento del tono simpático producido por la acción central de la insulina, la activación del sistema renina-angiotensina, la disfunción endotelial y expresión de la ET-1 o la acción vasoconstrictora provocada por los ácidos grasos libres.
A este cuarteto se ha añadido un estado disfibrinolítico imputable a la presencia de niveles elevados del inhibidor del activador del plasminógeno (PAI-1), producidos por las células endoteliales y musculares lisas en respuesta al estímulo de los TG y la insulina. Esta respuesta es más intensa en los pacientes con el polimorfismo del PAI-168,70.
Sustrato del receptor de la insulina (IRS-1)
La proteína IRS-1 (insuline receptor substrate) actúa de intermediaria entre el receptor de la insulina y la cascada de señales que determinan la incorporación a la membrana de la proteína GLUT-4 (el canal de transporte en cuya ausencia la membrana es impenetrable a la glucosa). La causa de la resistencia a la insulina es con toda probabilidad una menor actividad de la IRS72. Los estudios epidemiológicos ya conocían que la resistencia a la insulina y la hiperinsulinemia aumentan notablemente el riesgo de infarto de miocardio, aunque el paciente no fuera diabético74, por lo que al describirse el polimorfismo G972R del gen IRS-1 se incluyó enseguida en la lista de posibles factores de riesgo genético. En esta mutación se produce un cambio de arginina por glicina en el codón 972 dando lugar a dos alelos G y R. La prevalencia en la población general de los portadores de la mutación, es decir, del genotipo GR (el RR sería excepcional) es del 6-7%71. Estudios experimentales y clínicos han confirmado la asociación de este polimorfismo con el deterioro de la función de la proteína IRS, con la resistencia a la insulina y la hiperinsulinemia, y el aumento del riesgo coronario. La mutación se encuentra con mayor frecuencia en los enfermos con lesiones angiográficas (19 frente al 6-7%). Cabe subrayar que el riesgo se multiplica por 7 si el paciente es obeso y por 27 si coexiste además con datos clínicos del síndrome metabólico71.
GENES RELACIONADOS CON LA FUNCION ENDOTELIAL
El descubrimiento de que la disfunción endotelial puede promover tanto la formación de las placas de ateroma como la aparición de accidentes coronarios ha sido una de las aportaciones más relevantes de los últimos años para la comprensión de la génesis de la aterosclerosis. El endotelio, aparte de su actividad vasodilatadora macro y microvascular, tiene también una importante acción antitrombótica, antiproliferativa, antiapoptótica y, en particular, una función antioxidante que resulta decisiva para proteger la pared vascular contra el efecto aterogénico de las LDL oxidadas y otros agentes lesivos. Todas estas funciones estan íntimamente relacionadas con la capacidad del endotelio de producir óxido nítrico (NO), que contrarresta la acción de los radicales libres de oxígeno (aniones superóxido, O2-) liberados en el metabolismo tisular75. En condiciones normales la producción de NO predomina sobre la generación de aniones superóxido y mantiene bajo control el estrés oxidativo. Si la producción de NO es insuficiente o la generación de radicales libres de oxígeno es excesiva, puede romperse este equilibrio, y el predominio de los fenómenos oxidativos provoca la disfunción endotelial con todas sus consecuencias vasomotoras y metabólicas (fig. 5).

Fig. 5. Genes que modulan la función endotelial. Las flechas paralelas horizontales señalan el equilibrio entre la producción de radicales libres de oxígeno (O2-) por la NADH oxidasa y de óxido nítrico (NO) por la eNOS, que está bajo el control de la angiontensina II y la bradicinina (flechas convergentes). Los genes que codifican las enzimas se indican en cursiva y dentro de un recuadro. Las flechas divergentes 1, 2 y 3 indican los efectos desfavorables de O2- sobre: a) las células musculares lisas (potencian la producción de ET-1 y factores de crecimiento); b) la oxidación de las LDL, y c) la células endoteliales (que liberan citocinas proinflamatorias y elementos protrombóticos. En los cuatro recuadros de la parte inferior se señalan las alteraciones asociadas con la disfunción endotelial.
NADH/NADPH oxidasa
La fuente principal de producción de radicales libres de oxígeno son las enzimas oxidativas, entre las que destaca la NADH/NAFDPH oxidasa, la xantinoxidasa, la mieloperoxidasa, las 15-lipooxigenasa o la ciclooxigenasa. Se ha descrito un polimorfismo C242T del gen del p22phox, uno de los elementos del transporte de electrones de la NADH oxidorreductasa microsómica en las células musculares lisas, que disminuye la producción de anión superóxido en la pared vascular y mejora la función del endotelio coronario, por lo que se sospecha que puede reducir la sensibilidad a la enfermedad coronaria76,77.
Sintasa del NO
La capacidad de contrarrestar el estrés oxidativo y mantener la integridad de la función endotelial puede depender de un polimorfismo de la sintasa endotelial del NO, en el que se produce un cambio de G →T en la posición que corresponde al nucleótido 894 (Glu 298 Asp), el polimorfismo NOS3 G894T. En pacientes que requieren tratamiento vasoconstrictor durante la cirugía cardíaca, se ha comprobado que la respuesta vascular vasopresora a la fenilefrina (estimulación alfaadrenérgica) es más intensa en portadores del alelo 894 T (TT y GT) que en los homozigotos GG, lo que indica que la capacidad vasodilatadora está deprimida a causa de la menor producción de NO78. Esta menor actividad de la enzima NOS3 podría ser un factor de riesgo coronario de primera magnitud79, sobre todo en pacientes con angina de pecho vasoespástica80.
Metilentetrahidrofolatorreductasa (MTHFR), hiperhomocisteinemia y folatos
La hiperhomocisteinemia se considera hoy un importante factor de riesgo coronario, cerebrovascular y periférico. La elevación de sólo 5 mmol/l (el límite superior normal es de 15 μmol/l) aumenta el riesgo coronario en un 60 y 80% en varones y mujeres, respectivamente. Se trata de una alteración frecuente (ocurre en el 5-7% de la población general) a la que se atribuye un papel causal decisivo en el 10% de los infartos de miocardio en EE.UU. El efecto aterogénico se debe a que se acumula en las células endoteliales, aumenta el estrés o xidativo y provoca la disfunción endotelial4,81.
En la mitad de los casos, la causa de la hiperhomocisteinemia es la presencia del genotipo homozigoto TT del polimorfismo C677T de la MTHFR (5,10 metilentetrahidrofolatorreductasa), que codifica una variante termolábil de la enzima MTHFR, menos eficiente en su función de degradación de la homocisteína a metionina y su eliminación de las células. En estos casos, la cifra de homocisteína plasmática aumenta en un 25%, aunque esta elevación al parecer sólo ocurre en presencia de una relativa deficiencia de folatos. La enzima es dependiente de folato y la enzima mutada tiene una menor capacidad de utilización de folatos (y por tanto el requerimiento dietético es mayor); pero la homocisteinemia puede ser normal si la ingesta de folatos es suficiente81. La prevalencia de este polimorfismo es del 10-15% de la población general. Otras mutaciones de las enzimas que intervienen en el metabolismo de la metionina, como la sintasa de la citationa, son más raras4.
La segunda causa de la hiperhomocisteinemia es la ingesta inadecuada de cofactores vitamínios como el ácido fólico y las vitaminas B6 y B12. Lo común es que coincidan el factor ambiental y el genético81. Otras causas son el hipotiroidismo, la insuficiencia renal, el efecto de determinados medicamentos como los anticonvulsionantes, el ácido nicotínico, el metrotexato, el alcohol y sobre todo el tabaco, que interfieren con la síntesis de la vitamina B6. Éste es un nuevo mecanismo aterogénico a añadir a los efectos perniciosos del tabaco.
Varios estudios han descrito que el genotipo homozigoto TT aumenta el riesgo coronario, especialmente si el nivel de folatos es bajo81; pero no faltan resultados contradictorios. Se ha publicado un metaanálisis de 23 estudios con 6.000 casos y 6.000 controles que no pudo comprobar ninguna asociación entre el genotipo y la enfermedad coronaria82. Sin embargo, un metaanálisis holandés más reciente, sin embargo, con cerca de 5.000 casos, refiere un resultado ligeramente positivo, y describe un aumento gradual del riesgo coronario en los genotipos heterozigoto y homozigoto del alelo T83. Es posible que la confirmación definitiva requiera estudios en poblaciones de gran tamaño, o poblaciones vulnerables con carencia de folatos.
Las hipercisteinemias graves, con concentraciones plasmáticas de hasta 100-400 μmol/l, capaces de producir homocisteinuria, son enfermedades mendelianas excepcionales, como la deficiencia homozigota de la CBS, que, aparte de las deformaciones esqueléticas y el retraso mental, se asocia a una aterosclerosis prematura grave, con complicaciones tromboembólicas arteriales y venosas antes de los 30 años4.
GENES RELACIONADOS CON LA RESPUESTA INFLAMATORIA
La aterosclerosis se considera un proceso inflamatorio crónico que aparece como respuesta reparadora de una lesión vascular metabólica, física o ambiental. La gravedad y persistencia de esta respuesta inflamatoria local podría estar determinada genéticamente, ya sea a través de genes que influyen en la capacidad de producción de los mediadores inflamatorios (citocinas, moléculas de adhesión) o la capacidad de respuesta de los monocitos y linfocitos a los estímulos inflamatorios, tales como las LDL oxidadas, los radicales libres de O2- y las endotoxinas o lipopolisacáridos (LPS).
Por otra parte, cada vez hay más indicios de que algunos procesos inflamatorios o infecciosos sistémicos pueden favorecer la aparición de la enfermedad coronaria y sus complicaciones. Tal es el caso de la respuesta inflamatoria crónica de baja intensidad que se detecta en clínica por el aumento de los niveles de los mediadores inflamatorios (la proteína C reactiva [PCR], amiloidosis A, interleucina 6 [IL-6], fibrinógeno [FGN], sICAM), aunque no está claro si esta actividad inflamatoria es causa o consecuencia de la actividad de la placa. Una infección que ha despertado gran interés es la causada por Chlamydia pneumoniae, uno de los agentes patógenos respiratorios más frecuentes, cuya asociación con la enfermedad coronaria está respaldada por numerosos estudios seroepidemiológicos y experimentales.
Entre los polimorfismos que pueden influir en la respuesta inflamatoria local o sistémica destacan los siguientes84-87:
Interleucina 6 (IL-6)
La IL-6 ocupa un papel central en la génesis de la reacción inflamatoria sistémica y la respuesta de la fase aguda, pues es la única citocina que puede estimular la síntesis de todas las demás proteínas que intervienen en la respuesta inflamatoria84. Se ha descubierto un polimorfismo, el G/C -174 del gen de la IL-6, que disminuye la capacidad de producción de IL-6 y puede amortiguar la respuesta inflamatoria. Los niveles circulantes basales de IL-6 en el genotipo CC son inferiores en un 50% a los del genotipo GG, y los estudios preliminares demuestran que el riesgo de infarto de miocardio se reduce a la mitad (riesgo relativo de 0,54). Se ha comprobado in vitro e in vivo que las variaciones en la producción de la IL-6 en respuesta a los estímulos de la LPS o IL-1 pueden dar origen a diferencias importantes en la producción de fibrinógeno, de moléculas de adhesión del endotelio o de la agregabilidad plaquetaria84.
Interleucina 1 (IL-1β)
También podrían influir en la progresión de la aterosclerosis los polimorfismos de la interleucina 1 (IL-1): el polimorfismo C/T -511 del gen de la IL-1β y el polimorfismo del antagonista del receptor R-IL-1 (aR-IL-1)85. Se ha descrito que los pacientes seropositivos a Chlamydia son más propensos a padecer infarto de miocardio si son portadores del genotipo IL-1 C/C y/o el alelo 2- o 3-repeat alelle del aR-IL-1. Al parecer el efecto aterogénico de la infección sólo se manifestaría en presencia de este polimorfismo, que confiere la susceptibilidad necesaria. Esto podría explicar los resultados a veces conflictivos de los estudios sobre la asociación entre Chlamydia y la enfermedad coronaria85.
Receptores de la quemocina CCR5 y del CD14 de los monocitos
Otro gen en estudio es el de los receptores de la quemocina CCR5. El polimorfismo D-CCR5 (deleción de 32 pb) puede tener cierto efecto protector contra el infarto de miocardio, atribuible a la atenuación de la respuesta inflamatoria y al enlentecimiento de la aterosclerosis86.
Se ha identificado también un polimorfismo, 260 C/T, en el promotor del gen de receptor CD14, que podría modificar la respuesta de los monocitos/macrófagos a los estímulos infecciosos y citocinas proinflamatorias, y en consecuencia podría aumentar su adhesividad al endotelio, su capacidad de infiltración y reclutamiento en la pared vascular y su transformación en macrófagos. Mediante citometría de flujo se ha podido comprobar que la densidad de los receptores CD14 de los monocitos está aumentada en los homozigotos TT y que este genotipo se encuentra con mayor frecuencia en pacientes que han padecido un infarto de miocardio87.
GENES RELACIONADOS CON LA TROMBOSIS
Trombosis y aterosclerosis estan íntimamente relacionadas entre sí. El aumento de la actividad procoagulante o la disminución de la capacidad fibrinolítica pueden contribuir al progreso de la placa de ateroma y/o favorecer la formación del trombo y la oclusión coronaria en el momento de la rotura.
Gen del fibrinógeno y tabaco
El fibrinógeno se ha revelado como un factor predictivo muy potente de enfermedad coronaria, tanto como pueden serlo los factores de riesgo clásicos. Este hecho refuerza la importancia de su papel en la progresión de las estenosis coronarias y/o en la aparición de las complicaciones. Los mecanismos en los que interviene son múltiples: a) es el precursor de la fibrina, que se deposita en la placa de ateroma y favorece su crecimiento a través de la organización del trombo; b) estimula la proliferación de las células musculares lisas; c) aumenta la viscosidad de la sangre y tiene un efecto reológico sobre la función endotelial; d) interviene en los fenómenos de adhesión, y e) además, puede influir en la gravedad de los accidentes coronarios agudos, porque los trombos formados con niveles altos de fibrinógeno son más resistentes a la lisis que los formados con niveles bajos7.
Se ha comprobado reiteradamente que la concentración del fibrinógeno depende en parte de su genotipo88-90. La molécula está formada por dos subunidades idénticas (proteína dimérica), cada una de las cuales consta de tres cadenas polipéptidas (α, β, γ), codificadas en tándem por tres genes distintos del brazo largo del cromosoma 4. Se han descrito varios polimorfismos íntimamente asociados en el gen de la cadena beta (FNG B), entre los que destacan el polimorfismo -455 G/A (el polimorfismo guanina/adepolimorfismo RFLP), y el polimorfismo Bcl I.
En el estudio europeo ECTIM, los portadores del alelo -455 A, que representan el 20-25% de la población general, tenían una cifra de fibrinógeno más alta (351 mg/dl) que los del genotipo GG (338 mg/dl). La cifra era de 388 mg/dl en el genotipo AA88. Las diferencias en otros estudios oscilan entre el 1 y el 10%3.
Una de las características más notables de este polimorfismo es su importante interacción con el tabaco, pues el aumento de la concentración del fibrinógeno únicamente se observa (o es más aparente) en fumadores. El tabaco es el principal determinante ambiental del fibrinógeno plasmático, y su supresión lo reduce en un 10%4. Este efecto se atribuye a que la síntesis hepática del fibrinógeno forma parte de la respuesta de la fase aguda mediada por las citocinas, pues el gen tiene una región del promotor sensible al estímulo de la IL-6. El tabaco promueve la síntesis hepática a través de su importante efecto proinflamatorio estimulante de la producción de IL-64,88. Este efecto podría acentuarse por la coexistencia del polimorfismo 148 C/T FGN B, localizado en la región promotora sensible a la IL-6. La coexistencia de estos dos polimorfismos explicaría por qué algunos fumadores presentan cifras tan elevadas de fibrinógeno88.
A pesar de que la relación entre el genotipo y el fibrinógeno (fenotipo intermedio) se ha comprobado repetidamente88-90, los estudios de asociación con la enfermedad coronaria angiográfica o la incidencia de infarto de miocardio (fenotipo clínico) no han sido todo lo convincentes que cabría esperar. La mayoría son negativos. De todos modos se ha descrito su asociación con la enfermedad vascular periférica en el Edinburgh Artery Study4,89, con el infarto de miocardio en la rama francesa del ECTIM88, y con el infarto familiar en una población seleccionada del GISSI-290.
Polimorfismos de los factores VII y XIII
El factor VII interviene junto con el factor tisular en el inicio de la cascada de la coagulación (vía extrínseca). Dos polimorfismos pueden tener un importante papel protector contra el infarto de miocardio, debido a que se asocian a una notable disminución de los niveles plasmáticos del factor VII y de la propensión a la trombosis. Se trata del polimorfismo FVII R353Q, con sustitución de arginina (R) por glutamina (Q) en el codón 353 del exón 8, y del polimorfismo 5'F7R, que incluye la inserción de 10 pb en la posición -323 de la región 5' del promotor, con dos alelos, A1 (sin inserción) y A2 (con inserción). Los individuos con el genotipo homozigoto QQ o A2A2 tienen unos niveles plasmáticos del factor VIIa (activado) inferiores en un 70% al de los homozigotos RR o A1A1. Esta importante reducción tiene al parecer una repercusión clínica significativa, pues el alelo Q o el A2 se asocia a una reducción del riesgo de infarto de miocardio a la mitad. La mayor protección la confiere el genotipo A2A2, que reduce el riesgo en un 70% comparado con el genotipo A1A191. Así se explicaría por qué algunos pacientes no sufren un infarto de miocardio a pesar de que padecen una aterosclerosis coronaria grave91,92. Otros estudios, sin embargo, niegan cualquier asociación93.
La contribución del polimorfismo Val34Leu del factor XIII es más discutible, aunque se ha descrito un efecto protector contra el infarto y la trombosis venosa94,95. Este factor interviene en la fase final de la coagulación estabilizando el coágulo de fibrina y confiere una mayor resistencia frente a la fibrinólisis.
Se ha apuntado que también podrían estar implicados en la enfermedad coronaria los polimorfismos del factor VIII, que interviene, junto con el factor IX (vía intrínseca), en la propagación del coágulo, o del factor de Von Willebrand (vWF), la molécula más decisiva en la interacción de las plaquetas y pared vascular4.
Gen del PAI-1 y triglicéridos
El aumento de la cifra plasmática del PAI-1 (inhibidor del activador del plasminógeno) se considera un importante factor de riesgo trombótico y aterogénico, ya que amortigua la actividad fibrinolítica natural del t-PA. Esta proteína está bajo el control del polimorfismo 4G/5G del gen PAI-1, que se caracteriza por la presencia de 5 nucleótidos de guanina en la zona promotora en vez de cuatro. El alelo 5G reduce la actividad transcripcional y la producción de fibrinógeno, por lo que en principio puede considerarse un cambio favorable. Los portadores del genotipo homozigoto 4G4G tienen cifras de PAI-1 superiores en un 10-50% a las del 5G/5G, y una capacidad fibrinolítica del plasma reducida. Como se desprende de los estudios de asociación, los portadores del alelo 4G serían más propensos a padecer la enfermedad coronaria96-98, aunque las conclusiones del ECTIM97 han sido negativas. Un metaanálisis reciente demuestra una débil propensión del alelo 4G al infarto de miocardio98.
Cabe destacar la existencia de una relación muy significativa entre las concentraciones plasmáticas de los TG (y la obesidad visceral) y del PAI-1. Los individuos con niveles altos de TG y genotipo 4G/4G tienen cifras de PAI-1 superiores a las de los portadores del genotipo 5G/5G (véase síndrome metabólico). Se ha identificado en la región promotora del gen una secuencia sensible a los TG que podría explicar esta interacción genética/ambiental.
Gen del t-PA
La capacidad del endotelio de liberar grandes cantidades de t-PA en los accidentes coronarios agudos para contrarrestar el depósito masivo de fibrina parece estar influida por el polimorfismo Alu I del gen del
t-PA (inserción que contiene un sitio de restricción para la enzima Alu I). En el estudio de Rotterdam se ha comprobado su asociación con el infarto de miocardio, que está pendiente de confirmar99.
Otras variantes genéticas
Las enfermedades monogénicas producidas por mutaciones del sistema hemostático aumentan el riesgo de hemorragia o de trombosis (generalmente venosa), pero no tienen al parecer efectos notables sobre la aterosclerosis. Tal es el caso de las enfermedades hemorrágicas causadas por un defecto del vWF, la proteína indispensable para la formación del coágulo plaquetario, y la hemofilia A (deficiencia del factor VIII ligado al cromosoma X), que dificulta la formación del trombo de fibrina. Aunque se ha sospechado que pueden tener un efecto protector, no son una garantía contra la aterosclerosis. Por otra parte, las variaciones genéticas trombofílicas, como las del factor V de Leiden (presente en el 5% de la población) y el A20210G del gen de la protrombina (en el 2%), aumentan el riesgo de trombosis venosa profunda pero no parecen influir en el riesgo de trombosis arterial o de infarto4-6.
POLIMORFISMOS PLAQUETARIOS
Entre las variaciones genéticas que pueden modificar la actividad de las plaquetas destacan los polimorfismos de las tres glucoproteínas principales (GP) de la membrana plaquetaria: la GP IIb/IIIa, el receptor del fibrinógeno; la GP Ib-IX-V, el receptor de vWF; y el GP Ia-IIa, el receptor del colágeno100-103.
Polimorfismo PlA1 /PlA2 del gen de la glucoproteína IIIa
El gen IIIa codifica uno de los componentes del receptor IIb/IIIa, el receptor que se une al FGN y da origen a la agregación de las plaquetas y al trombo plaquetario. Se conoce la existencia de un polimorfismo de la glucoproteína IIIa, en el que la sustitución T/C en la posición 1565 da lugar a dos variantes, los antígenos plaquetarios 1 y 2 (PlA1 y PlA2).
El Framingham Offspring Study ha podido confirmar que el polimorfismo GP IIIa PlA2 aumenta muy significativamente la agregabilidad plaquetaria101, lo que indica que este alelo, presente en el 16% de la población, puede ser un factor de riesgo genético importante. Algunos estudios lo han asociado con el infarto de miocardio, los síndromes coronarios agudos, el riesgo de trombosis del stent o de complicaciones isquémicas en las intervenciones percutáneas. Otros han puesto en duda cualquier relación101,102.
Polimorfismo del gen GP Ib (polimorfismo Kozak)
El receptor GP Ib-IX-V cumple un papel crucial en la adhesión de las plaquetas a la pared vascular lesionada en la fase inicial de la hemostasia, gracias a que se une a la matriz subendotelial rica en vWF. El polimorfismo -5 T/C en la secuencia Kozak de la GP Iba (la subunidad del complejo GP Ib-IX-V que se une al vWF) se ha asociado a un aumento de la densidad del receptor GP Ib-IX-V en la membrana plaquetaria, lo que podría ser un factor de riesgo de trombosis plaquetaria en los síndromes coronarios agudos o las complicaciones postangioplastia coronaria transluminal percutánea. Los portadores del alelo -5C (portadores del polimorfismo Kozak) presentan angina inestable con más frecuencia que los del alelo natural (5T). La determinación del genotipo podría facilitar la estratificación del riesgo y el diseño del tratamiento antiplaquetario más idóneo103.
Polimorfismo del gen de la glucoproteína Ia
La GP Ia-IIa es el receptor de colágeno que estabiliza las plaquetas adheridas a la pared vascular. Algunos polimorfismos del gen GP Ia aumentan hasta 10 veces la densidad de los receptores de colágeno en la superficie de las plaquetas. Los estudios sobre su predisposición a los síndromes coronarios agudos han dado resultados dispares100.
IMPLICACIONES CLINICAS
El concepto de que la variabilidad genética individual puede desempeñar un papel determinante del riesgo coronario resulta muy atractivo, sobre todo si se tiene en cuenta que los factores de riesgo clásico no son capaces de justificar más del 30-50% de los casos de enfermedad coronaria4. Se han descrito ya numerosos alelos de riesgo y la lista de genes candidatos no cesa de ampliarse. Nuestro conocimiento, sin embargo, adolece de importantes limitaciones. En la mayoría de los casos la asociación con el riesgo coronario o el fenotipo intermedio es muy modesta o está en entredicho. A pesar de sus limitaciones, está tomando cuerpo la hipótesis de que el riesgo de padecer la enfermedad coronaria es función del número de polimorfismos desfavorables que porta un individuo. Las posibles consecuencias de este concepto son importantes:
1. El análisis del perfil genómico (diagnóstico molecular) puede facilitar el diagnóstico de la susceptibilidad genética individual a partir de la suma de los alelos de riesgo presentes. Un ejemplo de lo que puede ser la realidad inmediata lo ilustra un reciente estudio preliminar en el que se analiza el riesgo en función de los 8 polimorfismos más relevantes identificados en el estudio de regresión, relacionados con los lípidos (LPL Pvu y Hind III, CETP), con el sistema renina-angiotensina-aldosterona (AGT, CMA y sintasa de la aldosterona) y la trombosis (PAI-1)62. La lista definitiva de factores genéticos quizá tenga que esperar a que el Proyecto Genoma Humano-2 (1998-2003) ofrezca el catálogo completo de polimorfismos y dispongamos de tecnología adecuada (microchips) para realizar estudios de asociación en muchos miles de individuos.
2. La descripción de las bases genéticas (fisiopatología) puede resolver el problema de la clasificación etiológica de la aterosclerosis coronaria, una vieja aspiración.
3. Los tests genéticos pueden ser de utilidad en la predicción de la eficacia de un fármaco en un paciente determinado (farmacogenómica). El genotipo es un factor a tener muy cuenta al interpretar los resultados de los ensayos clínicos.
4. Un tema distinto es considerar el posible beneficio de la estratificación precoz del riesgo genético mediante pruebas del ADN (el cribado sistemático). En qué medida puede sustituir o completar el diagnóstico bioquímico tradicional es todavía tema de debate.
5. El objetivo más ambicioso de la medicina preventiva, la modificación genética que evite el infarto (la terapia génica), queda más lejos, pero quizá menos de lo que parece.
Parcialmente financiado con una ayuda del FIS 00/0284.
Correspondencia: Prof. F. Navarro López.
Servicio de Cardiología (ICMCV). Hospital Clínico.
Villarroel, 179. 08036 Barcelona.
Correo electrónico: navarro@medicina.ub.es