

Los bloqueadores beta son moléculas ampliamente utilizadas y capaces de antagonizar los receptores adrenérgicos (RA) beta, pertenecen a la familia de receptores acoplados a proteínas G y reciben el estímulo de las catecolaminas endógenas. Tras su estimulación, se activan cascadas intracelulares que en última instancia originan la contracción cardiaca o la dilatación vascular, según el subtipo y su ubicación. Se han descrito 3 subtipos, que se expresan de manera diferenciada en el organismo (RA-β1, β2 y β3), y el subtipo β1 es el más abundante en el corazón. Desde su descubrimiento, los RA-β se han convertido en diana para combatir las enfermedades cardiovasculares. Desde su invención por James Black a finales de los años cincuenta, los bloqueadores beta han supuesto una revolución en la terapia cardiovascular. Hasta ahora se dispone de 3 generaciones: los bloqueadores beta no selectivos, los bloqueadores beta cardioselectivos (antagonista selectivo de β1) y los bloqueadores beta vasodilatadores. Estos constituyen la tercera generación y son capaces de bloquear los β1 además de tener actividad vasodilatadora, bien bloqueando los RA-α1 o activando los RA-β3. Los bloqueadores beta todavía se utilizan ampliamente en la clínica tras más de 50 años desde la introducción del propranolol en el mercado por su capacidad para reducir la frecuencia cardiaca y, por lo tanto, la demanda miocárdica de oxígeno en el caso de una angina.
Palabras clave
Desde un punto de vista farmacológico clásico, los bloqueadores beta son antagonistas de los receptores adrenérgicos (RA) β, que desempeñan un papel importante en el control de procesos fisiológicos, tales como la presión arterial, la frecuencia cardiaca y la resistencia de las vías respiratorias o reactividad, así como otros procesos metabólicos y del sistema nervioso central1–4. Tras su descubrimiento por el ganador del premio Nobel sir Henry H. Dale en 19065 (figura 1), los RA se convirtieron en dianas clave en enfermedades cardiovasculares, tales como la hipertensión y la insuficiencia cardiaca (IC), en enfermedades respiratorias como el asma y otras enfermedades no menos importantes, como la hipertrofia prostática benigna, la congestión nasal, la obesidad y el dolor, entre muchas otras1–4.

No obstante, no fue hasta 1948 que Raymond P. Ahlquist observó 2 vías diferenciadas que inducían respuestas farmacológicas en función del órgano en el que se estudiaban los fármacos. A partir de estos experimentos, Ahlquist dividió los RA en 2 tipos, los RA-α (asociados con funciones mayormente «excitadoras» como la vasoconstricción) y los RA-β (asociados con funciones mayormente «inhibidoras» como la vasodilatación y un efecto «excitador», la estimulación del miocardio)6. Más tarde, en 1958, sir James Black introdujo el primer bloqueador beta en la búsqueda de un tratamiento capaz de reducir el consumo de oxígeno en el caso de aparición de una angina de pecho y corroboró la teoría de Ahlquist. Esta invención, considerada uno de los logros más importantes en medicina en el siglo xx, le valió a Black y el mundo de los RA un segundo premio Nobel en 19887 (figura 1).
En 1967, Alonzo M. Lands et al. propusieron dividir los RA-β en 2 subtipos distintos: los RA-β1, principalmente presentes en el corazón, y los RA-β2, encargados de la relajación vascular y de las vías respiratorias8. Esta clasificación fue respaldada por el posterior descubrimiento de los antagonistas selectivos de los RA-β19. En seguida se identificó un tercer subtipo, con tantas semejanzas como diferencias e insensible a los fármacos utilizados con mayor frecuencia, en las células del tejido adiposo pardo de ratas, al que se denominó RA-β310,11.
El logro más reciente es el conseguido por Robert J. Lefkowitz y Brian K. Kobilka, quienes contribuyeron a identificar la interacción de los RA-β con estructuras celulares, su regulación dinámica y desensibilización y, por último, a solucionar la estructura cristalina tridimensional de los RA-β2 en 2007 (figura 1). Esta investigación llevó a Lefkowitz y Kobilka a recibir el tercer premio Nobel a un trabajo sobre los RA en 201212.
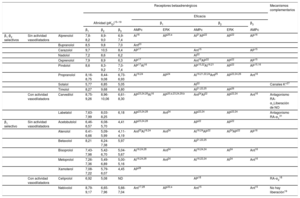
HISTORIA, DESARROLLO Y CLASIFICACIÓN DE LOS BLOQUEADORES BETAEn 1958, sir James Black tuvo la brillante idea de apuntar a una reducción en la demanda miocárdica de oxígeno, en lugar de a un aumento en su disponibilidad por vasodilatación, en el caso de una angina de pecho. Inspirado por la teoría de Ahlquist, la obsesión de Black fue hallar un fármaco que fuera capaz de bloquear el efecto «excitador» del miocardio atribuido al RA-β, que controlaría así la frecuencia cardiaca. Mientras, los Laboratorios Eli Lilly lanzaron el dicloroisoproterenol, que se creía que era un broncodilatador, pero se vio que tenía cierto efecto antagonista en el corazón13. Cuando Black tuvo conocimiento de ello, se le ocurrió la idea de sintetizar análogos del dicloroisoproterenol que pudieran ser más potentes y selectivos en sus propiedades de bloqueo betaadrenérgico. En esta búsqueda, inventó el primer bloqueador beta aprobado para su uso clínico, el propranolol14. El propranolol es el prototipo de los bloqueadores beta de primera generación, que son fármacos con afinidades parecidas por los RA-β1 y β2 (tabla 1)15–32, y por este motivo se los considera «bloqueadores beta no selectivos». Dentro de este grupo, el propranolol es el fármaco con mayor experiencia clínica acumulada y que se ha indicado más veces33 (tabla 2).
Clasificación y mecanismos de acción de los bloqueadores beta
| Receptores betaadrenérgicos | Mecanismos complementarios | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Eficacia | |||||||||||
| Afinidad (pKD)15–19 | β1 | β2 | β3 | ||||||||
| β1 | β2 | β3 | AMPc | ERK | AMPc | ERK | AMPc | ||||
| β1-β2 selectivos | Sin actividad vasodilatadora | Alprenolol | 7,8-8,2 | 8,9-9,0 | 6,9-7,4 | AI16 | AP20,a | AI21AP22 | AP22 | AP16 | |
| Bupranolol | 8,5 | 9,8 | 7,0 | Ant23 | |||||||
| Carazolol | 9,7 | 10,5 | 8,4 | AP17 | Ant15 | AP15 | |||||
| Nadolol | 7,2 | 8,6 | 6,2 | AI22 | |||||||
| Oxprenolol | 7,9 | 8,9 | 6,3 | AP17 | Ant15AP22 | AP22 | AP15 | ||||
| Pindolol | 8,6 | 8,3-9,2 | 7,0-7,4 | AP17AI16 | AP15,22AI16,21 | AP22 | AP15,16 | ||||
| Propranolol | 8,16-8,75 | 8,44-9,08 | 6,73-6,93 | AI16,24 | AP24 | AI16,21,22,24Ant25 | AP22,24,26 | Ant16 | |||
| Sotalol | 5,77 | 6,85 | 5,05 | AI22 | Canales K+27 | ||||||
| Timolol | 8,27 | 9,68 | 6,80 | AI21,22,25 | AP26 | ||||||
| Con actividad vasodilatadora | Carvedilol | 8,75-9,26 | 8,96-10,06 | 6,61-8,30 | AP23,24,28AI16 | AP20,a,23,24,29,b | Ant24AI22 | AP22,24 | Ant16 | Antagonismo RA-α1Liberación de NO | |
| Labetalol | 7,63-7,99 | 8,03-8,25 | 6,18 | AP23,24,28 | Ant24 | AP22,24 | AP22,24 | Antagonismo RA-α131 | |||
| β1 selectivo | Sin actividad vasodilatadora | Acetobutolol | 6,46-6,57 | 6,08-5,70 | 4,41 | AP23,24,28 | AP22 | AP22 | |||
| Atenolol | 6.41-6,66 | 5,09-5,99 | 4,11-4,19 | Ant23AI16,24 | Ant24 | AI16,24AP22 | AI24AP22 | AP16 | |||
| Betaxolol | 8,21 | 6,24-7,38 | 5,97 | AI21,22,25 | |||||||
| Bisoprolol | 7,43-7,98 | 5,42-6,70 | 5,04-5,67 | AI16,24,28 | Ant24 | AI16,24,24 | AI24 | Ant16 | |||
| Metoprolol | 7,26-7,36 | 5,49-6,89 | 5,00-5,16 | AI16,24,28 | Ant24 | AI16,22,24 | AI24 | Ant16 | |||
| Xamoterol | 7,08-7,22 | 5,79-6,07 | 4,45 | AP28 | |||||||
| Con actividad vasodilatadora | Celiprolol | 6,92 | 5,08 | ND | AP18 | RA-α218 | |||||
| Nebivolol | 8,79-9,17 | 6,65-7,96 | 5,66-7,04 | Ant17,28 | AP32,a | Ant15 | Ant15 | No hay liberación19 | |||
AI: agonismo inverso; AMPc: monofosfato de adenosina cíclico; Ant: antagonismo; AP: agonismo parcial; ERK: cinasa regulada por señales extracelulares; ND: no determinado; pKD: –log de la concentración del fármaco que se une al 50% de la población de receptores (constante que expresa afinidad); RA: receptor adrenérgico.
Indicaciones más habituales de los bloqueadores beta
| β1-β2 selectivos | β1 selectivos | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sin actividad vasodilatadora | Con actividad vasodilatadora | Sin actividad vasodilatadora | Con actividad vasodilatadora | |||||||||
| Insuficiencia cardiaca | Carvedilol | Bisoprolol | Metoprolol | Nebivolol | ||||||||
| Hipertensión | Propranolol | Nadolol | Carvedilol | Labetalol | Atenolol | Bisoprolol | Metoprolol | Celiprolol | Nebivolol | |||
| Hipertensión ocular | Timelol | Betxolol | ||||||||||
| Enfermedad cardiaca isquémica | Propranolol | Nadolol | Carvedilol | Atenolol | Bisoprolol | Metoprolol | Celiprolol | |||||
| Arritmia | Propranolol | Nadolol | Sotalol | Atenolol | Metoprolol | |||||||
| Hemorragia portal hipertensiva (profilaxis) | Propranolol | Carvedilol | ||||||||||
| Migraña (profilaxis) | Propranolol | Nadolol | Metoprolol | |||||||||
| Tirotoxicosis | Propranolol | Metoprolol | ||||||||||
| Feocromocitoma | Propranolol | |||||||||||
| Temblor esencial | Propranolol | |||||||||||
| Ansiedad | Propranolol | |||||||||||
Pocos años más tarde, en 1966, en la búsqueda de derivados capaces de evitar el efecto broncodilatador del propranolol en pacientes con asma (debido a su actividad antagonista β2), el equipo de Imperial Chemical Industries lanzó el practolol, el primer compuesto representativo de los bloqueadores beta de segunda generación, que son fármacos que muestran una mayor afinidad por el RA-β1 que por el β2 y se consideran bloqueadores beta «β1 selectivos» o «bloqueadores beta cardioselectivos» por la presencia predominante del subtipo β1 en el corazón. En 1975, el practolol se retiró del mercado y el posterior curso de desarrollo de fármacos dio origen a los nuevos bloqueadores beta cardioselectivos. Los fármacos más representativos de este grupo son el atenolol y el metoprolol34,35 (tabla 1 y tabla 2).
Los bloqueadores beta de tercera generación son fármacos con propiedades vasodilatadoras adicionales y, por esta característica, se denominaron «bloqueadores beta vasodilatadores». Esta actividad vasodilatadora es beneficiosa porque reduce la resistencia vascular periférica al tiempo que mantiene o mejora el gasto cardiaco, el volumen sistólico y la función del ventrículo izquierdo. Los compuestos que pertenecen a este grupo pueden ser selectivos o no selectivos para el RA-β1, pero muestran mecanismos adicionales, como la actividad antagonista del RA-β1 (carvedilol y labetalol) y la liberación de óxido nítrico (NO) (nebivolol), que explica su actividad vasodilatadora. Por otra parte, los bloqueadores beta vasodilatadores tienen efectos neutros (labetalol y nebivolol) o beneficiosos (carvedilol) en el metabolismo de la glucosa y los lípidos, mientras que la mayoría de los estudios clínicos indican que los bloqueadores no vasodilatadores tienden a un efecto negativo en los parámetros de glucosa y lípidos36 (tabla 1 y tabla 2).
Este campo emergente se ha completado con formas farmacéuticas de acción prolongada y acción ultracorta, que han contribuido a la mejora del arsenal terapéutico34,35.
Hoy no hay ninguna duda de que la introducción de los bloqueadores beta hace más de 50 años revolucionó la farmacoterapia humana y tuvo un impacto positivo en la salud de millones de personas con enfermedades cardiovasculares y no cardiovasculares.
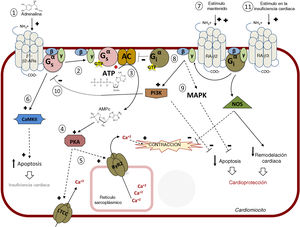
RECEPTORES BETAADRENÉRGICOS CARDIACOS: VÍAS DE SEÑALIZACIÓN Y MODULACIÓNEs fundamental conocer mejor el funcionamiento de las complejas redes de señalización desencadenadas por la estimulación del RA-β y sus alteraciones en situaciones patológicas para comprender los efectos de los bloqueadores beta y para el diseño de estrategias terapéuticas. Hay tres subtipos de RA-β (RA-β1, RA-β2 y RA-β3) en el tejido cardiaco. Si bien todos los RA-β pertenecen a la superfamilia de los receptores de membrana acoplados a proteínas G (GPCR) y comparten varias características estructurales y funcionales, los tres subtipos muestran afinidades distintas por determinados ligandos, una expresión celular específica y patrones de localización subcelular también específicos, acoplamiento diferencial en las cascadas de señalización y distintos mecanismos reguladores2,3,37 (figura 2).
![Vía intracelular mediada por receptores adrenérgicos β (RA-β). 1. Vía principal: las catecolaminas se unen a los RA-β e inducen el acoplamiento a la proteína Gs heterotrimérica. 2. Disociación de la subunidad Gαs-GTP y activación de la adenilil ciclasa (AC). 3. Síntesis de monofosfato de adenosina cíclico (AMPc). 4. Activación de la proteincinasa A (PKA). 5. Fosforilación coordinada por la PKA de varias dianas, como el canal de calcio de tipo L (CCTL) de la membrana plasmática o el canal de calcio RyR2 del retículo sarcoplásmico; el resultado es un aumento de la concentración citosólica de Ca2+ disponible para la contracción del músculo cardiaco. 6. La estimulación continua (tal como se describe en la insuficiencia cardiaca crónica) del RA-β1 induce la apoptosis por medio de la proteincinasa II dependiente de Ca2+/calmodulina (CaMKII) causante de apoptosis y afección cardiaca. 7. La estimulación continua de β2 (aumentada cuando se utilizan bloqueadores β1 selectivos) induce el acoplamiento a la proteína Gi. 8. La AC es inhibida por la subunidad Gα-GTP de Gi. 9. La subunidad Gβγ de Gi induce tanto la inhibición de la apoptosis (por medio de la estimulación de proteincinasas activadas por mitógenos [MAPK] como la vía del fosfatidilinositol 3-cinasa [PI3K]-proteincinasa B y los efectos deletéreos mediados por Gs (10), que llevan a la cardioprotección. 11. En la insuficiencia cardiaca, la estimulación del RA-β podría conducir a cardioprotección y a una reducción del remodelado cardiaco por medio de la activación de la sintasa del óxido nítrico (NOS). Adaptado con permiso de Watson et al.37.](https://static.elsevier.es/multimedia/03008932/0000007200000010/v1_201909250714/S0300893219301666/v1_201909250714/es/main.assets/gr2.jpeg?xkr=eyJpdiI6IlhvT2xaUm1GQmFyeGRHOEI2NmUrMVE9PSIsInZhbHVlIjoiNmpaVE1haTIzMXduOXFkK2xNbnpHQmU1VDNHNGZTTDBpOVhDTUhKL1pWYzRadHlFanREUWFBSjNacnU0RDM5NUg2ZlYyc2V2a2F0UkpOSVUxNFZXMDc4RTdWVkJLTXJlSEo2b3JCSjNoWmpESy9yZXZvTkxlRHlsRWVZZkZjQVA5Nlg5YUhGMEtXYWwrR0ZaWGRLNEFIYWUwd21xNWMyV3hvbk9tT0ZXWi9SM1lyN25Sako0NEpFTXlQZ29idWNwNmtPU0N6SS9jUFRGaHhadVJqa3c0ekNpYzFNSWl6a3AwcVNnUXVPUUt4RFJtQXZTYnZUWVZ0UTNxUzRTWmVsS0ZiZlBiTFpDU2I1RGtGQUdMczMwNGQ2TDc3eXRxTW5GeUxPN1hrUzcwZWs9IiwibWFjIjoiZDM0NTEwZmY4ZTFiODE4NDEzM2UyZjhmNWE4Nzg3NGU2YWU5YzY4NjRlZWMzZTI3ZGI4NGUxMzcwMjEyMDg2ZSIsInRhZyI6IiJ9 "Vía intracelular mediada por receptores adrenérgicos β (RA-β). 1. Vía principal: las catecolaminas se unen a los RA-β e inducen el acoplamiento a la proteína Gs heterotrimérica. 2. Disociación de la subunidad Gαs-GTP y activación de la adenilil ciclasa (AC). 3. Síntesis de monofosfato de adenosina cíclico (AMPc). 4. Activación de la proteincinasa A (PKA). 5. Fosforilación coordinada por la PKA de varias dianas, como el canal de calcio de tipo L (CCTL) de la membrana plasmática o el canal de calcio RyR2 del retículo sarcoplásmico; el resultado es un aumento de la concentración citosólica de Ca2+ disponible para la contracción del músculo cardiaco. 6. La estimulación continua (tal como se describe en la insuficiencia cardiaca crónica) del RA-β1 induce la apoptosis por medio de la proteincinasa II dependiente de Ca2+/calmodulina (CaMKII) causante de apoptosis y afección cardiaca. 7. La estimulación continua de β2 (aumentada cuando se utilizan bloqueadores β1 selectivos) induce el acoplamiento a la proteína Gi. 8. La AC es inhibida por la subunidad Gα-GTP de Gi. 9. La subunidad Gβγ de Gi induce tanto la inhibición de la apoptosis (por medio de la estimulación de proteincinasas activadas por mitógenos [MAPK] como la vía del fosfatidilinositol 3-cinasa [PI3K]-proteincinasa B y los efectos deletéreos mediados por Gs (10), que llevan a la cardioprotección. 11. En la insuficiencia cardiaca, la estimulación del RA-β podría conducir a cardioprotección y a una reducción del remodelado cardiaco por medio de la activación de la sintasa del óxido nítrico (NOS). Adaptado con permiso de Watson et al.37.")
Vía intracelular mediada por receptores adrenérgicos β (RA-β). 1. Vía principal: las catecolaminas se unen a los RA-β e inducen el acoplamiento a la proteína Gs heterotrimérica. 2. Disociación de la subunidad Gαs-GTP y activación de la adenilil ciclasa (AC). 3. Síntesis de monofosfato de adenosina cíclico (AMPc). 4. Activación de la proteincinasa A (PKA). 5. Fosforilación coordinada por la PKA de varias dianas, como el canal de calcio de tipo L (CCTL) de la membrana plasmática o el canal de calcio RyR2 del retículo sarcoplásmico; el resultado es un aumento de la concentración citosólica de Ca2+ disponible para la contracción del músculo cardiaco. 6. La estimulación continua (tal como se describe en la insuficiencia cardiaca crónica) del RA-β1 induce la apoptosis por medio de la proteincinasa II dependiente de Ca2+/calmodulina (CaMKII) causante de apoptosis y afección cardiaca. 7. La estimulación continua de β2 (aumentada cuando se utilizan bloqueadores β1 selectivos) induce el acoplamiento a la proteína Gi. 8. La AC es inhibida por la subunidad Gα-GTP de Gi. 9. La subunidad Gβγ de Gi induce tanto la inhibición de la apoptosis (por medio de la estimulación de proteincinasas activadas por mitógenos [MAPK] como la vía del fosfatidilinositol 3-cinasa [PI3K]-proteincinasa B y los efectos deletéreos mediados por Gs (10), que llevan a la cardioprotección. 11. En la insuficiencia cardiaca, la estimulación del RA-β podría conducir a cardioprotección y a una reducción del remodelado cardiaco por medio de la activación de la sintasa del óxido nítrico (NOS). Adaptado con permiso de Watson et al.37.
Ante la unión agonista, los GPCR se acoplan a proteínas G heterotriméricas, lo que facilita así el intercambio de GDP por GTP en las subunidades Gα, que por consiguiente se disocian de los dímeros βγ. Las subunidades libres Gα y βγ interactúan de forma transitoria con efectores (tales como adenilil ciclasas o fosfolipasas, entre otros) para desencadenar cascadas de transducción de señales4. Además, los GPCR activados por agonistas son fosforilados de manera específica en el tercer bucle citoplásmico o en el extremo C-terminal por cinasas de los GPCR (GRK), una familia de 7 proteincinasas de serina/treonina38,39. A continuación, las arrestinas β, proteínas citosólicas, son captadas por el receptor fosforilado, lo que lleva a que no se produzca el acoplamiento con la proteína G, en un proceso llamado desensibilización de los GPCR. Además, las arrestinas β pueden actuar como un armazón para las proteínas de la maquinaria endocítica y para muchos otros componentes asociados en la transducción de señales, lo que desencadena así la interiorización y el reciclaje del receptor mediado por clatrina y una segunda ola de cascadas de transducción independientes de la proteína G40. En consecuencia, los efectos generales de la estimulación de los GPCR serían resultado del equilibrio entre las ramas de la señalización de los GPCR dependientes de proteína G y las dependientes de GRK/arrestina β.
Eje de señalización β-adrenérgico/Gαs/PKALos RA-β del miocardio modulan la contractilidad y la relajación cardiacas a través de la fosforilación mediada por la proteincinasa A (PKA) de una variedad de proteínas que controlan el Ca2+ y elementos miofilamentosos. En condiciones fisiológicas, estos efectos involucran mayormente a los RA-β1 y los RA-β2, ya que estos receptores se expresan predominantemente en los cardiomiocitos humanos sanos (en proporción RA-β1:RA-β2 de 4:1), con escasa expresión del RA-β32,3. Curiosamente, en miocitos ventriculares de ratones, al parecer los RA-β1 se hallan presentes en todos los cardiomiocitos, mientras que el RA-β2 y el RA-β3 se detectan únicamente en el 5% de los miocitos pero son abundantes en las células del endotelio cardiaco, donde a su vez el RA-β1 se expresa poco41, lo que indica una integración heterogénea de la señalización del subtipo de RA-β en distintas células cardiacas (figura 2).
Tanto el RA-β1 como el RA-β2 pueden acoplarse a la proteína Gs. La activación de la subunidad Gαs lleva a la activación de la adenilil ciclasa (AC), que a su vez cataliza la formación de monofosfato de adenosina cíclico (AMPc) a partir de trifosfato de adenosina (ATP). Las AC 5 y 6, que pueden activarse por medio de la Gαs y desactivarse por medio de la Gαi y calcio, son las isoformas predominantes de la AC en el corazón42. El aumento local de AMPc desencadena la activación de PKA por la unión de sus subunidades reguladoras, con lo que se libera la subunidad catalítica funcional, que fosforila de forma coordinada una variedad de sustratos en distintas localizaciones celulares. La fosforilación del canal de calcio de tipo L (CCTL) de la membrana plasmática aumenta la entrada de Ca2+, que a su vez activa el receptor de rianodina 2 (RyR2) en la membrana del retículo sarcoplásmico (RS) a través de un mecanismo denominado liberación de Ca2+ inducida por Ca2+, que resulta en un aumento de la concentración citosólica de Ca2+ disponible para la contracción (figura 2). Este proceso de liberación diastólica de Ca2+ en el RS se refuerza después bien por la fosforilación directa de RYR2 mediada por PKA, bien por la estimulación indirecta de la calcio-calmodulina cinasa II (CaMKII) de este canal del RS. Al mismo tiempo, la fosforilación de la troponina cardiaca I y de la proteína C de unión a la miosina cardiaca facilita el acoplamiento excitación-contracción. Por otro lado, la PKA fosforila e inhibe el fosfolambán, un inhibidor de la ATPasa de Ca2+ del RS, lo que acelera la reabsorción de Ca2+ citoplásmico en el RS y da cuenta de la relajación. Además de estos efectos inotrópicos y lusitrópicos, la estimulación adrenérgica también fomenta la modulación directa del AMPc de los canales regulados por nucleótidos cíclicos activados por hiperpolarización que transportan la corriente de marcapasos, que eleva la frecuencia cardiaca (efecto cronotrópico)42-44.
Merece la pena observar que los RA-β y sus vías efectoras dirigidas a las proteínas que controlan el Ca2+ se hallan muy compartimentalizadas en los cardiomiocitos. La señalización del RA-β2 tiene una localización más local, ya que estos receptores se encuentran preferentemente en los túbulos T, donde se ubican junto con el CCTL en caveolas, mientras que el RA-β1 se distribuye de manera generalizada por los túbulos T y el sarcolema y genera señales de AMPc que se propagan por toda la célula45. Asimismo, las proteínas del armazón denominadas proteínas de anclaje a la cinasa A contribuyen a unir complejos proteínicos tales como AC, PKA, sustratos y fosfodiesterasas en compartimentos subcelulares concretos que permiten la regulación espaciotemporal de la señalización por AMPc42.
Junto a estos efectos generales, otras dianas del eje RA-β/AMPc/PKA pueden contribuir a la respuesta celular generalizada. La activación adrenérgica de la PKA desencadena mecanismos inhibidores de retroalimentación4. Tanto los RA-β1 como los RA-β2 comprenden secuencias de consenso para la fosforilación de la PKA, y ello disminuye la afinidad de estos receptores por la Gαs, lo que lleva a la desensibilización. La fosforilación mediada por PKA de los RA-β cardiacos también induce la captación de fosfodiesterasa 4 del AMPc en las proximidades de los receptores, lo que fomenta la degradación local del AMPc en estimulación prolongada del receptor. Por otro lado, la fosforilación de la PKA del RA-β2 favorece el acoplamiento del receptor a Gαi, que contribuye a inhibir posteriormente la producción de AMPc a través de la AC y que también desencadena vías de señalización alternativas, tales como la cascada Gβγ/PI3K/proteincinasa B (Akt)3. Además de controlar la homeostasis equilibrada del AMPc, la fosforilación del RA-β1 por parte de la PKA facilita su interacción con 14-3-3ɛ y, por lo tanto, aleja esta proteína de los canales Kv11.1, reguladores fundamentales de la repolarización y la refractariedad cardiacas46, mientras que la PKA también puede estimular la cascada Akt/sintasa del NO endotelial (eNOS)/monofosfato de guanosina cíclico (GMPc)/proteincinasa G (PKG), lo cual conduce a la inactivación del CCTL y a una entrada reducida de Ca2+ extracelular44.
Vías dependientes de la arrestina βLas GRK y la arrestina desempeñan un papel muy importante en la regulación y la señalización del RA-β cardiaco. La GRK2 y la GRK5 se expresan en la mayoría de las células cardiacas, mientras que la GRK3 solo se encuentra en los cardiomiocitos. La estimulación agonista fomenta de modo secuencial la fosforilación mediada por GRK de los RA-β y la captación de arrestinas β (en el corazón humano la arrestina β1 es más abundante que la arrestina β2), lo que lleva a la finalización de la señalización de proteína G, a la interiorización del receptor y a la regulación por disminución47,48. Además, las arrestinas β inician cascadas de señalización independientemente de la activación de las proteínas G, tal como la activación de la cascada de cinasas regulada por señales extracelulares (ERK) vía interacción con c-Src o transactivación del receptor del factor de crecimiento epidérmico (EGFR) en la fosforilación del RA-β1 por parte de la GRK53,49. Se ha señalado que esta última vía es cardioprotectora, tal como indicaban la apoptosis aumentada y la dilatación cardiaca en ratones transgénicos que sobreexpresan un RA-β1 mutante que carece de sitios de fosforilación GRK y, en consecuencia, incapaces de captar arrestina β y transactivar el EGFR49. Un módulo de señalización RA-β1/arrestina β también estimula el procesamiento de micro-ARN cardiacos protectores tales como los miR-150 y otros, que protegen el corazón del ratón de una lesión isquémica50. Curiosamente, se ha visto que el bloqueador beta carvedilol, además de bloquear la hiperactivación perjudicial de proteína G, actúa como un ligando del RA-β sesgado para la arrestina β, capaz de desencadenar tales vías adaptativas mediadas por la arrestina β20,29,50, cosa que abre vías interesantes de investigación en relación con mecanismos de acción diferenciales de los bloqueadores beta.
Cascadas de transducción dependientes de proteínas Epac desencadenadas por los RA-β cardiacosAdemás de la PKA, hay nuevas pruebas que indican que otro efector del AMPc, la proteína de intercambio activada por AMPc denominada Epac, también desempeña un papel importante en la función y la enfermedad cardiacas relacionadas con los RA-β. La formación de AMPc mediada por el RA- β1 activa la Epac1, que a su vez activa las sintasas neuronales de NO y CaMKII a través de PI3K y Akt, lo que favorece así la fuga de calcio del RS por medio de la fosforilación de RYR44,51. El «señalosoma» Epac1 se halla muy compartimentalizado, lo que puede contribuir a las diferencias funcionales entre los subtipos cardiacos de RA-β.
Características alteradas de la señalización del RA-β en situaciones patológicasLa hiperactividad del sistema nervioso simpático y la concentración aumentada de catecolaminas circulantes son mecanismos compensatorios tempranos desencadenados en respuesta a la afección miocárdica y la disfunción para mantener el gasto cardiaco a través de efectos de mediación betaadrenérgica en la contractilidad. No obstante, tal activación crónica de los RA-β fomenta una serie de alteraciones en las redes de señalización cardiaca (como la desregulación y la desensibilización excesiva del RA-β y la funcionalidad/expresión alterada de las GRK, las arrestinas β y las proteínas Epac), que contribuyen en última instancia a que se produzca una remodelación cardiaca patológica, hipertrofia ventricular, fibrosis, arritmia e IC2,3.
La estimulación crónica del RA-β está asociada a apoptosis celular y a pérdida de funcionalidad de la bomba. La regulación por disminución selectiva de la expresión de RA-β1 altera la ratio fisiológica entre el RA-β1 y el RA-β2, que se convierte en el principal subtipo de RA-β durante la evolución de la IC52. Además, en este contexto, la localización del RA-β2 normal se redistribuye desde los túbulos transversos hasta una cresta celular global y gana una amplia distribución, que lleva a una señal más difusa del AMPc53. En los cardiomiocitos deteriorados, la activación persistente del RA-β2 también fomenta cascadas dependientes de CaMKII causantes de la aparición de hipertrofia, apoptosis, disfunción cardiaca y arritmias por medio de la sobrecarga de Ca2+ del RS54 (figura 2). La inactivación redox del RA-β155, la dosis aumentada de Epac1 prohipertrófica51 o de Gαi, las concentraciones alteradas o el estado de S-nitrosilación de las arrestinas β56 también pueden contribuir a la alteración en la señalización del RA-β1 y el RA-β2 en contextos patológicos, así como los autoanticuerpos anti-RA-β1 presentes en algunos pacientes con miocardiopatía dilatada idiopática57. Por otro lado, parecería que el RA-β3 (que es menos susceptible a la desensibilización, puede acoplarse a ambas proteínas Gs y Gi y también fomenta la estimulación del eje eNOS/NO/GMPc/PKG) no se ha modificado o incluso que se halla regulado por aumento en contextos patológicos. Se ha señalado que el bloqueo del RA-β1 por parte del metoprolol regula por aumento los RA-β3, lo cual provoca la activación de la señalización cardioprotectora de la esfingosina-1-fosfato58, aunque existen datos contradictorios sobre el papel beneficioso de los agonistas del RA-β3 en la IC2.
También cabe señalar que se ha observado que la expresión aumentada de GRK2 en pacientes y modelos de experimentación de IC debida a hipertensión o isquemia crónicas y se ha visto que su ablación genética o inhibición son cardioprotectoras en modelos animales52. El aumento de GRK2 puede ayudar en un inicio al miocardio a contrarrestar el sobreimpulso del RA-β y reducir el riesgo de taquiarritmia, pero a la larga no se produce un buen ajuste, lo que resulta en la desensibilización y regulación por disminución del RA-β y la contractilidad defectuosa. La dosis aumentada de GRK2 cardiaco también altera la función mitocondrial, compromete la biodisponibilidad del NO y fomenta la resistencia de la insulina cardiaca, que favorece en última instancia la remodelación del miocardio deteriorado y la evolución a IC39,59,60.
La GRK2 también emerge como un elemento clave para conectar la insulina cardiaca con las cascadas de RA-β en condiciones patológicas, puesto que esta cinasa puede regularse por aumento ya sea mediante catecolaminas o con una dieta rica en grasas, y puede modular tanto la señalización del RA-β como de la insulina59,61-64. Curiosamente, se ha visto que algunos bloqueadores beta, así como el ejercicio, reducen la concentración de GRK2 en el miocardio2,47, lo cual puede contribuir a los efectos beneficiosos de estos fármacos.
Señalización del RA-β en otros tipos de células cardiacasAunque la mayoría de los estudios se han centrado en el papel de la señalización adrenérgica en los cardiomiocitos, esta también puede desempeñar un papel fisiopatológico muy importante en otros tipos de células cardiacas3. En fibroblastos, la activación de los RA-β2, pero no de los RA-β1, fomenta la degradación de colágeno, la autofagia, la activación de ERK y la proliferación celular a través de la transactivación del EGFR65,66. En las células endoteliales, la estimulación del RA-β2 activa la eNOS y la vasodilatación. Por último, el RA-β1 y otros RA-β también se presentan como moduladores relevantes del tránsito de leucocitos hasta el corazón lesionado, un proceso clave para la remodelación y la reparación cardiacas posteriores a una lesión cardiaca67. El impacto funcional integrado de los RA-β y los bloqueadores beta en los distintos tipos de células es fundamental para la investigación futura.
MECANISMO DE ACCIÓN DE LOS BLOQUEADORES BETALa afinidad es la capacidad de un fármaco para unirse al receptor, y la eficacia es la capacidad de inducir una respuesta. Los fármacos se clasifican como agonistas o antagonistas dependiendo de si son eficaces o no.
Todos los bloqueadores beta comparten un mismo mecanismo, que es su afinidad para unirse a los RA-β aunque, al contrario que los agonistas de los RA-β, no son eficaces a la hora de producir respuestas fisiológicas. Los bloqueadores beta compiten con agonistas por el sitio de unión al RA-β y la consecuencia es la inhibición de la actividad agonista. Por este motivo, clásicamente se han considerado antagonistas competitivos y sus efectos pueden contrarrestarse aumentando la concentración del agonista15.
A pesar de este mecanismo común, en estudios clínicos, los bloqueadores beta no se comportan como una clase única de fármacos. Por ejemplo, se ha probado que el bisoprolol, el carvedilol, el metoprolol y el nebivolol son útiles en el tratamiento de la IC, el bucindolol no tuvo ningún efecto positivo y el xamoterol aumentó la mortalidad15. Así pues, se requiere un análisis más riguroso del mecanismo de acción para comprender la utilidad clínica de este grupo.
Hay algunos aspectos que marcan la diferencia:
- •
Afinidad selectiva por los subtipos de RA-β. Los subtipos de RA-β no son entidades intercambiables y los bloqueadores beta muestran una afinidad distinta por cada uno de los subtipos de RA-β, lo que resulta en un perfil farmacológico concreto.
Las consecuencias funcionales del bloqueo del RA-β1 en el corazón son bradicardia y mejora del tiempo de llenado coronario durante la diástole, disminución de la demanda de oxígeno y reducción de la renina; todos estos efectos son beneficiosos en la IC y la isquemia miocárdica68. No obstante, las consecuencias del bloqueo de los RA-β2 o β3 no son positivas, ya que se impide la broncodilatación mediada por el subtipo β2, así como los mecanismos cardioprotector y vasodilatador desencadenados por ambos subtipos. De hecho, en los vasos, estos se hallan presentes en las células vasculares del músculo liso, así como en el endotelio, donde se acoplan a la vía eNOS/NO-GMPc/PKG, que facilita la vasodilatación69.
El primer grupo de bloqueadores beta clínicamente disponibles mostraba mayor afinidad por los subtipos β1 y β2 que por el subtipo β3 (tabla 1), por lo que, a dosis clínicas, su actividad terapéutica se relacionaría principalmente con el bloqueo del RA-β1 y el RA-β2 (tabla 2).
Los bloqueadores beta «cardioselectivos» tienen mayor afinidad por el subtipo β1 que por los subtipos β2 y β3. Cuando se administran a dosis bajas, inhiben los RA-β1 cardiacos, pero no la vasodilatación o broncodilatación mediada por el RA-β2. Sin embargo, la selectividad del RA-β1 es relativa (tabla 1) y se pierde a dosis más altas y, en consecuencia, el uso de bloqueadores β1 selectivos debería considerarse con precaución en pacientes con enfermedades de las vías respiratorias.
También cabe destacar que, entre los bloqueadores beta aprobados para el tratamiento de la IC, el bisoprolol y el nebivolol son los más β1 selectivos, el metoprolol muestra selectividad β1 moderada y el carvedilol, selectividad β2 escasa (tabla 1). Así pues, no es posible determinar si la selectividad β1 es esencial para unos resultados positivos máximos en la IC.
- •
Agonismo inverso. La teoría tradicional sobre la interacción entre receptor y fármaco se basa en una población quiescente de receptores que solo actúan cuando se unen a un ligando con eficacia (agonista). No obstante, se sabe que los RA-β, en ausencia de agonistas, pueden adoptar de forma espontánea conformaciones activas capaces de regular sistemas de señalización70 y acoplamiento a distintos mecanismos transductores3. En consecuencia, hay que revisar la interpretación simplista de que los bloqueadores beta son fármacos sin eficacia para activar el receptor.
La evidencia de esta «actividad constitutiva» de los RA-β en ausencia de agonistas llevó al descubrimiento de fármacos que podían reducirla. Puesto que los efectos de estos fármacos eran opuestos a los de los agonistas, se los consideró «agonistas inversos»71, es decir, en lugar de ocupar únicamente el sitio de unión y bloquear así las acciones de los agonistas, estabilizan las conformaciones del receptor que no están acopladas a proteínas G y evitan las vías de señalización activadas de forma constitutiva. Aunque inicialmente esta idea se vio con escepticismo, hoy día se acepta que todos los receptores pueden señalizar en ausencia de agonistas y la mayor parte de bloqueadores beta caracterizados anteriormente como antagonistas se consideran ahora agonistas inversos16,21,23. ¿Cuál es la importancia de esta observación? En un sistema con actividad constitutiva cuantificable, un agonista inverso reducirá la respuesta del receptor, mientras que un antagonista no lo hará, aunque ambos impiden la actividad agonista.
Además, la actividad constitutiva del receptor resulta en la activación de los mecanismos de desensibilización que provocan la regulación por disminución de los receptores70. El tratamiento con un agonista inverso detiene esta regulación por disminución, lo cual aumenta la expresión de los receptores y mejora la sensibilidad a la estimulación agonista71. La exposición sostenida del RA-β2 humano a agonistas inversos hizo que se duplicara (aproximadamente) en la membrana la concentración del receptor, mientras que el tratamiento equivalente con un antagonista no sirvió para producir este efecto72.
Los estudios con modelos humanos y animales muestran regulación por aumento de los RA-β1 y β2 en el corazón o de los RA-β2 en los linfocitos con el tratamiento crónico con propranolol, lo que explica la hipersensibilidad observada del RA-β tras la retirada súbita de propranolol15. Por otro lado, bloqueadores β1 selectivos tales como el atenolol, el metoprolol y el bisoprolol aumentan la densidad del RA-β1, pero no la del β215. Puesto que una característica general de los pacientes con IC es una disminución de la densidad del RA-β1 cardiaco52,73, la regulación por aumento debería servir para restablecer las respuestas contráctiles máximas. No obstante, el carvedilol no sirvió para regular por aumento los RA-β cardiacos en pacientes con IC, pero fue tan efectivo como el metoprolol y el bisoprolol en la mejora del funcionamiento cardiaco15. Así pues, es aún objeto de debate si la regulación por aumento del RA-β por parte de los bloqueadores beta podría ser una propiedad beneficiosa.
En la tabla 1 se sintetizan los datos sobre la actividad agonista inversa de los bloqueadores beta.
- •
Agonismo parcial. Tradicionalmente, se ha considerado que algunos bloqueadores beta tienen actividad simpaticomimética intrínseca. Esta actividad aparece si el fármaco tiene actividad antagonista en el subtipo de RA-β1, pero se comporta como un agonista en otro u otros subtipos, o si el fármaco puede fomentar una respuesta parcial de 1, 2 o los 3 subtipos (agonista parcial). La consecuencia de la activación parcial de los RA-β es el bloqueo de la actividad estimuladora de agonistas de alta eficacia, tales como las catecolaminas, pero la estimulación de un bajo nivel de respuesta del RA-β en ausencia de un agonista. Esta acción combinada podría ser beneficiosa, ya que se manifiesta únicamente cuando el sistema simpático se activa74. No obstante, los bloqueadores beta con actividad agonista parcial en los RA-β1 son al parecer menos beneficiosos en el tratamiento de la IC28. Por otro lado, una actividad antagonista en el RA-β1 junto con una actividad agonista en los RA-β2 o β3 produce vasodilatación y un efecto cardioprotector que podría representar un beneficio adicional75.
Estudios anteriores con bloqueadores beta que detectan actividad simpaticomimética intrínseca no diferencian entre estos mecanismos o el subtipo involucrado. Más recientemente, la actividad agonista parcial en cada subtipo de RA-β se ha estudiado de manera exhaustiva a nivel celular y tisular17,74. Los estudios con subtipos humanos de RA-β muestran diferencias que dependen de los bloqueadores beta y del subtipo estudiado: el oxprenolol, el carazolol, el pindolol y el nadolol tienen efectos agonistas parciales muy claros sobre los RA-β1 y β3, pero carecen de actividad intrínseca significativa sobre los RA-β217. El celiprolol se ha descrito como un antagonista del subtipo β1, pero como agonista parcial sobre los RA-β2 y β376.
En la tabla 1 se sintetizan algunos de los datos disponibles y en algunos casos se muestran resultados contradictorios. El nebivolol no facilita la acumulación de AMPc en células que expresan los subtipos humanos de RA-β17,18,28 y no relaja la vejiga urinaria en ratas, una respuesta típica mediada por el RA-β318, de modo que en estas condiciones no se comporta como un agonista parcial o total. No obstante, el nebivolol, a través de la activación del RA-β3, induce la vasodilatación mediada por NO77-79 con un efecto inotrópico negativo80 y protege contra la lesión por infarto de miocardio81. Además, reduce la resistencia vascular pulmonar y mejora el funcionamiento del ventrículo derecho en un modelo porcino de hipertensión pulmonar crónica82. Estos resultados contradictorios podrían resolverse si se supone que, dependiendo del tipo de célula donde se exprese el RA-β3, se activaron distintas vías de señalización. Esta hipótesis enlaza con el siguiente apartado, donde se trata el concepto de «agonismo sesgado».
- •
Agonismo sesgado. Un RA-β puede acoplarse no únicamente a una sino a distintas proteínas G, lo que lleva a complejos perfiles de señalización, tales como la acumulación de AMPc y la activación de proteincinasas activadas por mitógeno2. Por otro lado, en el caso de los subtipos β1 y β2, también se ha observado señalización independiente de proteína G principalmente a través de arrestinas β, que se encargan de la maquinaria de desensibilización/endocitosis y de la señalización no canónica a través de vías intracelulares como la vía mediada por ERK1/22,28.
Se han identificado ligandos que se unen a los RA-β y activan subgrupos diferenciados y específicos de estas vías de señalización. Se ha hecho referencia a este fenómeno como «propagación de estímulo dirigido por ligandos», «selectividad funcional» y «agonismo sesgado»70,83. Particularmente sorprendentes son los estudios que observan que algunos bloqueadores beta tienen eficacias opuestas hacia 2 vías de señalización distintas, lo que indica que la eficacia es un parámetro más complejo de lo que en un inicio se creía. De hecho, en teoría son posibles múltiples combinaciones de eficacia. Los compuestos podrían ser agonistas para las 2 vías, agonistas inversos para las 2 vías o tener eficacias opuestas en cada una. Por ejemplo, el propranolol, que actúa como agonista inverso sobre el RA-β2 con la vía Gs/AC/AMPc/PKA, se vio que era un agonista parcial cuando se probaba sobre la actividad de ERK24.
Y lo que es más curioso, entre un amplio grupo de bloqueadores beta, solo el carvedilol22 y el nebivolol32 indujeron la interiorización del RA-β2 y la activación de ERK1/2 independiente de proteína G, pero dependiente de arrestina β. Se han descrito resultados similares para el carvedilol, el alprenolol20 y el nebivolol32 en la transactivación del EGFR mediada por arrestina β y RA-β1. Puesto que esta transactivación del EGFR mediada por RA-β1 y arrestina β puede conferir cardioprotección49, los bloqueadores beta que activan esta vía podrían tener una eficacia superior en el tratamiento de los trastornos cardiovasculares20. Asimismo, el carvedilol fomenta de manera selectiva la captación y activación de Gαi en el subtipo de RA-β1 desencadenando la señalización mediada por arrestina β29.
No obstante, se recomienda precaución porque la célula o el estado fisiológico pueden llevar a resultados e interpretaciones distintos de los sistemas de señalización. Así pues, otros investigadores84 no fueron capaces de hallar pruebas de la captación de arrestina β por parte de estos bloqueadores beta que actúan sobre los RA-β2 (tabla 1).
- •
Mecanismos adicionales. Las propiedades de algunos bloqueadores beta son independientes de sus propiedades de bloqueo beta, pero contribuyen a su eficacia terapéutica. Se resumen en la tabla 1 e incluyen:
- –
Bloqueo del canal de K+, ejercido por el sotalol. Esta característica confiere al sotalol una actividad antiarrítmica adicional que se caracteriza por la ralentización de la repolarización y el potencial de acción prolongado en los tejidos cardiacos27.
- –
Actividad antagonista del RA-β1 ejercida por el carvedilol30 y el labetalol31. Esta acción adicional de bloqueo adrenérgico β1 conduce a vasodilatación con una reducción de la resistencia vascular periférica que actúa para mantener un mayor rendimiento del gasto cardiaco. Por contra, los bloqueadores beta no vasodilatadores tienden a aumentar la resistencia vascular periférica y reducir el gasto cardiaco y el funcionamiento del ventrículo izquierdo.
- –
Actividad de liberación de NO, en la que se halla implicado un efecto vasodilatador adicional. Esta propiedad se observó en el caso del nebivolol y podría estar mediada por una actividad agonista parcial, principalmente sobre el RA-β3, aunque no pueden descartarse otros mecanismos bien determinados85. La liberación aumentada de NO, acompañada de menor estrés oxidativo, lleva a un aumento de la biodisponibilidad de NO19 que participa en la actividad antihipertensora del nebivolol. De igual modo, el carvedilol aumenta considerablemente las concentraciones plasmáticas de NO por estimulación de la NOS86 y mejora la disponibilidad de NO derivada de sus propiedades antioxidantes. Sin embargo, no parece que estas acciones estén mediadas por una actividad agonista parcial sobre el RA-β3.85.
Desde su invención hace más de 50 años, los bloqueadores beta siguen siendo uno de los grupos de fármacos más útiles en la práctica clínica. Siguen utilizándose para su propósito original en el tratamiento de la enfermedad cardiaca isquémica, pero paradójicamente también son efectivos en la IC congestiva. Además, los bloqueadores beta también se utilizan como fármacos antihipertensores y en el tratamiento de las arritmias cardiacas, la hemorragia por varices esofágicas y la hipertensión pulmonar. Por otro lado, los bloqueadores beta tienen otras aplicaciones, como el tratamiento del glaucoma, los temblores, la migraña, la ansiedad y el hipertiroidismo. Cuanto más se conoce sobre sus mecanismos de acción intracelular específicos, mayor es el número de aplicaciones terapéuticas. Las nuevas vías de investigación deberían centrarse en el estudio detallado del mecanismo no explorado, específico del tipo de célula, de los bloqueadores beta a los que hay que considerar como moléculas individuales más que como un grupo homogéneo de fármacos. Medio siglo más tarde, los bloqueadores beta siguen sorprendiendo a la comunidad científica con nuevas aplicaciones terapéuticas que James Black nunca habría imaginado.
FINANCIACIÓNF. Mayor cuenta con financiación del Ministerio de Economía, Industria y Competitividad (MINECO) de España (subvención SAF2017-84125-R), del CIBERCV-Instituto de Salud Carlos III, España (subvención CB16/11/00278, cofinanciada con la contribución del Fondo Europeo de Desarrollo Regional) y del Programa de Actividades en Biomedicina de la Comunidad de Madrid-B2017/BMD-3671-INFLAMUNE. E. Oliver es receptor de una Ayuda del Programa de Atracción de Talento (2017-T1/BMD-5185) de la Comunidad de Madrid. El Centro Nacional de Investigaciones Cardiovasculares (CNIC) está financiado por el Ministerio de Ciencia, Innovación y Universidades y la Fundación Pro CNIC, y es un Centro de Excelencia Severo Ochoa (SEV-2015-0505). También se agradece el apoyo institucional del Centro de Biología Molecular Severo Ochoa de la Fundación Ramón Areces.
CONFLICTO DE INTERESESNo se declara ninguno.