
La hipertensión pulmonar se define por una presión media en la arteria pulmonar ≥ 25 mmHg. La hipertensión arterial pulmonar (HAP) es una forma caracterizada por una presión capilar pulmonar ≤ 15 mmHg y un aumento de las resistencias vasculares pulmonares (≥ 3 UW) que sin tratamiento conlleva mal pronóstico por insuficiencia del ventrículo derecho1. Entre sus etiologías, se incluyen la idiopática (HAPI), la hereditaria (HAPH), la inducida por fármacos o toxinas y la relacionada con otras causas como las cardiopatías congénitas (HAPC). Se denomina HAPH cuando se demuestra un patrón familiar o una mutación patogénica; el gen BMPR2 es el relacionado con más frecuencia, hasta en el 75% de los casos de HAPH y el 25% de los de HAPI1-3. Las mutaciones en este gen también podrían favorecer la aparición de HAP en algunas cardiopatías congénitas4.
Se realizó estudio genético y familiar de 8 casos de HAP seguidos en nuestro centro entre 2013 y 2016. Nuestro objetivo es analizar si sus resultados permitirían reclasificar a los pacientes. Se estudió mediante ultrasecuenciación masiva a los casos índice con un panel de 16 genes relacionados (ACVRL1, BMPR1B, BMPR2, CAV1, EIF2AK4, ENG, FOXF1, GDF2, KCNA5, KCNK3, NOTCH3, RASA1, SMAD1, SMAD4, SMAD9 y TOPBP1). Se consideraron patogénicas las variantes que se encontraban en una frecuencia alélica <0,01% en bases de datos públicas y que cumplían los criterios de patogenicidad establecidos5. Se realizaron árboles familiares y se ofreció cribado clínico y genético a los familiares de primer y segundo grado.
Las características de los pacientes y del estudio genético y familiar se detallan en la tabla. De los 8 casos índice estudiados, el estudio familiar permitió identificar 2 casos de HAPH al encontrarse antecedentes familiares de HAP. El estudio genético encontró variantes patogénicas o probablemente patogénicas relacionadas con HAP en 4 casos: 1 de HAPH, 2 de HAPI y 1 de HAPC (reclasificadas las últimas 3 como HAPH). Solo se detectaron mutaciones en el gen BMRP2, todas previamente descritas.
Características de los pacientes
| Caso índice | Sexo | Edad al inicio (años) | Tipo de HAP inicial | Tipo de HAP final | Otros hallazgos | Test vasodilatador | PAPm (mmHg) | RVP (UW) | Gen | Variante detectada | Descripción previa de la variante | Familiares | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Portador | Con HAP | ||||||||||||||
| V | M | V | M | ||||||||||||
| 1 | M | 36 | Hereditaria | Hereditaria | - | Negativo | 52 | 9,4 | BMPR2 | p.Cys34Phe | Probablemente patogénica | 2 | 0 | 0 | 3 |
| 2 | V | 10 | Asociada con cardiopatía congénita | Asociada con cardiopatía congénita y hereditaria | CIA ostium secundumpequeña | Negativo | 58 | 12 | BMPR2 | p.Arg491Trp | Patogénica | 0 | 0 | 0 | 0 |
| 3 | M | 41 | Idiopática | Hereditaria | Fístula arteriovenosa pulmonar <1 cmPosparto | Negativo | 59 | 28,5 | BMPR2 | p.Trp13* | Probablemente patogénica | 0 | 0 | 0 | 0 |
| 4 | M | 35 | Asociada con cardiopatía congénita | Asociada con cardiopatía congénita | CIA seno venoso superior amplia | Negativo | 48 | 6,9 | - | - | - | - | - | 0 | 0 |
| 5 | M | 34 | Idiopática | Idiopática | - | Positivo | 40 | - | - | - | - | - | - | 0 | 0 |
| 6 | M | 12 | Idiopática | Hereditaria | - | Negativo | 55 | 10,2 | BMPR2 | p.Asn442Thrfs*32 | Patogénica | 2 | 0 | 0 | 1 |
| 7 | M | 39 | Idiopática | Idiopática | Posparto | Positivo | 37 | 12,2 | - | - | - | - | - | 0 | 0 |
| 8 | M | 65 | Hereditaria | Hereditaria | Negativo | 49 | 9,3 | - | - | - | - | - | 0 | 1 | |
CIA: comunicación interauricular; HAP: hipertensión arterial pulmonar; M: mujer; PAPm: presión arterial pulmonar media; RVP: resistencia vascular pulmonar; V: varón.
Se realizó el cribado genético de 14 familiares de los pacientes con mutación identificada, cuyo resultado fue 1 mujer portadora afectada y 4 varones portadores sanos (tabla).
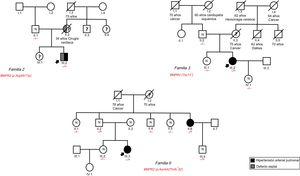
Las mutaciones identificadas fueron: a) caso 1: mujer, 36 años, clasificada como HAPH por haberse identificado a familiares de segundo grado con HAP; se encontró la mutación p.Cys34Phe previamente descrita en otra serie española6, lo que confirmaría su cosegregación; b) caso 2: varón, 10 años, con una comunicación interauricular pequeña y HAPC grave desproporcionada para el tamaño del defecto; se detectó la mutación p.Arg491Trp, y se interpretó como HAP de doble etiología: cardiopatía congénita incidental y hereditaria; su madre tenía una comunicación interventricular, falleció durante la cirugía correctora y posiblemente estaba afectada de HAP; no se pudo estudiar la rama materna porque no residía en España (figura); c) caso 3: mujer, 41 años, diagnosticada de HAPI, con una fístula arteriovenosa pulmonar <1 cm, sin otros hallazgos típicos de telangiectasia hemorrágica hereditaria; el gasto cardiaco se encontraba muy reducido, lo que indicaba que este hallazgo no tenía significación clínica; se detectó la mutación p.Trp13* en BMPR2, y no se encontró ninguna en los genes relacionados con la telangiectasia hemorrágica hereditaria, por lo que se reclasificó como HAPH; su padre no era portador, y no se pudo analizar su presencia en la rama materna (figura); d) caso 6: mujer, 12 años, con HAPI, en la que se detectó la mutación p. Asn442Thrfs*32, y se reclasificó como HAPH (figura).

Nuestros resultados son congruentes con lo publicado, y reflejan que el gen BMPR2 es el que se relacionado con HAP más frecuentemente, aunque se ha asociado con su aparición hasta 21 genes2. La penetrancia es mayor en mujeres y a menudo con inicio tras el parto debido a factores hormonales (casos 3 y 7) (tabla)1-3. Además, se ha visto que la respuesta vasodilatadora es más frecuente en la HAPI6 (casos 5 y 7) (tabla).
La guía de la Sociedad Europea de Cardiología indica la realización de estudio genético de los casos de HAPI, HAPH, telangiectasia hemorrágica hereditaria y enfermedad pulmonar venooclusiva1,2. No se considera su uso en la HAPC, aunque evidencia reciente muestra su utilidad en este contexto4. En nuestra serie, se incluyó a 2 pacientes con HAPC, y en 1 caso se detectó una mutación patogénica en BMPR2, lo que podría indicar su utilidad en este subgrupo, especialmente cuando la HAP es desproporcionada para el grado del defecto o cuando la HAP aparece tras repararlo.
Como conclusión, nuestros resultados muestran que el estudio genético y familiar en la HAP permite identificar las formas hereditarias, clasifica correctamente los diferentes tipos, incluidos los pacientes con HAPC con pequeños defectos. Aporta ventajas no solo en el tratamiento, sino también a la hora de ofrecer consejo genético a las familias, con la posibilidad de evitar la transmisión de la enfermedad a la descendencia, por lo que creemos que se debería considerar en la práctica clínica habitual.
FINANCIACIÓNCentro de Investigación Biomédica en Red (CIBERCV), Instituto de Salud Carlos III, CB16/11/00425. Parcialmente financiado por beca no condicionada de Actelion Pharmaceuticals.