
La observación de que «lo mismo no sirve para todos» en la prevención y el tratamiento de las enfermedades cardiovasculares, entre otras, ha propulsado el concepto de medicina de precisión. Su objetivo es proporcionar las mejores intervenciones basadas en la información adicional que aporta el genoma. El genoma humano se compone de miles de millones de pares de bases que contienen un código que controla cómo se expresan los genes. Este código depende de reguladores no estáticos que rodean el ADN y constituyen el epigenoma. Además, los factores ambientales también desempeñan un papel importante en esta compleja regulación. Se presenta una perspectiva general sobre los conceptos básicos de la biología molecular relacionados con la genética y la epigenética y un glosario de términos clave, se revisan varios ejemplos de polimorfismos y escalas de riesgo genético relacionadas con el riesgo cardiovascular, y se proporciona una visión general de los principales reguladores epigenéticos, como la metilación del ADN, las proteínas de unión a metilcitosina-fosfato-guanina, las modificaciones de histonas, otras regulaciones de histonas, los efectos de los microARN y otros reguladores emergentes. Otro desafío es entender cómo los factores ambientales (dieta, ejercicio, tabaco, etc.) pueden alterar el epigenoma y resultar en fenotipos saludables o no. Se comentan algunas interacciones entre gen y ambiente y se proporciona una visión metodológica general.
Palabras clave
Los avances tecnológicos acontecidos durante el Proyecto Genoma Humano y tras su finalización están permitiendo una gran reducción de costes y aumentando la inmediatez en el acceso a los resultados de los tests genéticos1. Ello ha posibilitado que se haya podido incorporar la determinación de marcadores genéticos a la investigación habitual en grandes cohortes de pacientes2,3 y en ensayos clínicos4,5. Los resultados de décadas de investigación en marcadores genéticos nos han permitido ahondar en el conocimiento sobre las bases moleculares de las principales enfermedades, entre ellas las enfermedades cardiovasculares y sus fenotipos intermedios6,7. Sin embargo, esta gran cantidad de información sobre los nuevos genes asociados con las distintas enfermedades, su posible modulación ambiental, etc., todavía sigue en un plano teórico y es necesario completar los siguientes pasos de validación diagnóstica, preventiva y/o terapéutica para que tenga lugar su traslación a la práctica clínica y a la población en general. Para acelerar este proceso, en Estados Unidos se puso en marcha el año 2015 una iniciativa denominada medicina de precisión8. Tal como en 2015 Collins y Varmus indicaban en su publicación sobre esta iniciativa8, el concepto de medicina de precisión —definido como «la implementación de estrategias de prevención y tratamiento que tienen en cuenta la variabilidad individual para optimizar el resultado»— no es nuevo, ya que en cierta forma se viene usando desde hace tiempo; como, por ejemplo, cuando se determina el grupo sanguíneo del paciente para guiar las transfusiones de sangre. Sin embargo, la novedad reside en que actualmente se dispone de una ingente cantidad de nuevos marcadores genómicos que pueden ayudar mejor a conocer el riesgo genético del paciente y predecir la respuesta interindividual a los tratamientos. Además de los marcadores genómicos basados en la secuencia del ADN, también están disponibles otros muchos biomarcadores ómicos (epigenómicos, transcriptómicos, metabolómicos, proteómicos, etc.) que, junto con la bioinformática y el desarrollo de nuevas herramientas computacionales para la gestión y la integración de estos datos, pueden proporcionar una información muy valiosa para mejorar drásticamente la prevención y el tratamiento de las enfermedades. Aunque inicialmente los esfuerzos de la medicina de precisión se han centrado fundamentalmente en el cáncer8, en el ámbito de las enfermedades cardiovasculares también se están produciendo avances que permitirán materializar los objetivos de esta nueva etapa de la medicina9.
Para que la medicina de precisión pueda pasar de la promesa a la realidad10, es necesario realizar ahora una intensa labor de investigación incorporando a los ensayos clínicos y otros estudios epidemiológicos la información de los marcadores ómicos y generar resultados que aporten un gran nivel de evidencia científica para guiar las decisiones en la nueva era8. Este paso requiere que los profesionales de la medicina y otras ciencias biomédicas adquieran una buena base de conocimientos en ómicas para así interpretar mejor y afrontar de manera más crítica los nuevos retos. Aunque el dogma central de la biología, propuesto por Francis Crick en 1958, indicaba que hay unidireccionalidad en el proceso de transmisión y expresión de la herencia genética, de manera que un ADN se transcribe como ARN mensajero y este se traduce como proteína, elemento que finalmente realiza la acción celular11, actualmente se sabe que no siempre es así y que existen importantes elementos reguladores que pueden hacer que el mismo ADN dé lugar a 2 o 3 proteínas distintas. Esto hace que se deba conocer mucho mejor no solo los elementos genéticos, sino los epigenéticos que contribuyen a esta regulación. Por ello, el objetivo de este trabajo es proporcionar una visión sucinta y actualizada de los conceptos básicos en biología molecular relacionados con la genética y la epigenética, para lo cual se presentan varios ejemplos de estudios realizados, aplicados fundamentalmente a las enfermedades cardiovasculares, y finalizar con una reflexión metodológica sobre las denominadas interacciones gen-ambiente. Otras revistas médicas como New England Journal of Medicine12,13 o Journal of the American Medical Association14–16, o más especializadas17–20, han publicado también varias revisiones sobre conceptos básicos de genética y epigenética que se aconseja consultar como complemento al contenido de esta revisión.
EL GENOMA HUMANOEl genoma humano tiene aproximadamente 6.000 millones de pares de bases (adenina, timina, guanina y citosina) de ADN y se organiza en 23 pares de cromosomas. Se estima que en el genoma humano hay unos 20.000-25.000 genes, muchos menos de los que se preveía en un principio12. A ello contribuye el hecho de que un mismo gen puede dar lugar a varias proteínas13, como se detalla más adelante. Debido a que cada par de bases en el ADN ocupa alrededor de 0,34 nm de longitud, cada célula diploide contendría aproximadamente 2 m de ADN si se extendiera. En total, el organismo humano sumaría 100.000 millones de metros de ADN21. Las histonas se encargan de compactar el ADN para que pueda caber en el microscópico núcleo celular. Las histonas son una familia de pequeñas proteínas cargadas positivamente y denominadas H1, H2A, H2B, H3 y H422. El ADN está cargado negativamente, debido a los grupos fosfato en su espina dorsal de fosfato-azúcar, por lo que las histonas se unen al ADN muy fuertemente. La unidad estructural básica y funcional de la cromatina es el nucleosoma, que contiene 8 proteínas histonas y aproximadamente 146 pares de bases de ADN. Los nucleosomas a su vez forman parte de otra estructura llamada cromatosoma, y cada cromatosoma empaqueta una media de 100 millones de pares de bases. Cada cromosoma es, por lo tanto, una larga cadena de nucleosomas21.
La cromatina a su vez se clasifica en eucromatina y heterocromatina. Hay diferencias de tinción, estructurales y funcionales entre ambas, pero se puede decir que en esencia la eucromatina es una estructura más laxa y transcripcionalmente activa que incluye la mayoría de los genes, mientras que la heterocromatina es más densa y contiene más secuencias repetidas como las que se puede encontrar en los telómeros. Estos son regiones altamente repetitivas que se encuentran al final de los cromosomas. La secuencia que más se repite en los telómeros humanos es la 5’TTAGGG 3’, pudiendo superar las 2.000 veces. Existe un complejo de 6 proteínas asociadas a los telómeros llamado shelterina o complejo protector, compuesto por TRF1 y TRF2, que a su vez interactúan con RAP1, TPP1, POT1 y TIN2 para asociarse al ADN telomérico22. La enzima encargada de la extensión de los telómeros se denomina telomerasa. Esta enzima, que es una retrotranscriptasa, mantiene la longitud de los telómeros; a partir de ARN como molde, añade la secuencia repetitiva d(TTAGGG) en el extremo 3’ del ADN telomérico, y proteínas accesorias específicas22. Varios estudios han asociado la longitud de los telómeros con distintas enfermedades23. En general se estima que unos telómeros más cortos se asociarían con mayor envejecimiento y más riesgo cardiovascular24.
Variaciones en la secuencia del genoma humanoLos genes constituyen la parte transcripcionalmente activa de los cromosomas. En la estructura de un gen se distinguen básicamente los intrones y los exones19. Los intrones son secuencias no codificantes, mientras que los exones son codificantes. Existen también unas regiones no codificantes al principio y final de cada gen llamadas 5’ región no traducida (UTR, por sus siglas en inglés) y 3’ UTR. La región inicial contiene el promotor del gen y la zona final se caracteriza por ser un lugar muy activo para la regulación por microARN. En la secuencia de ADN pueden producirse variaciones que pueden ser de distintos tipos (inserciones, deleciones, repeticiones, etc.)14,19. Las más conocidas son los cambios de un solo nucleótido, más conocidos por sus siglas en inglés, SNP (single nucleotide polymorphisms)14. Estos SNP pueden encontrarse en todas las zonas del gen. Si un polimorfismo se encuentra en un intrón, no afectará a la secuencia de aminoácidos de la proteína resultante y se dice que no es funcional. Sin embargo, actualmente se está comprendiendo mejor el significado de los polimorfismos en zonas no codificantes, ya que se puede afectar la funcionalidad del gen sin que ello implique cambio de aminoácido uniéndose o no otros reguladores, como se verá más adelante. Los polimorfismos que se encuentran en los exones pueden producir cambio de aminoácido o no, ya que algunas veces, aunque haya cambio de base, el triplete (grupo de 3 nucleótidos que determina un aminoácido) correspondiente que se genera sigue codificando el mismo aminoácido debido a que el código genético está «degenerado». Ello hace referencia a que, si se contabilizan las combinaciones de 4 elementos (las 4 bases del ADN) tomadas de 3 en 3, se contarían 64 aminoácidos distintos, pero solamente existen 20 aminoácidos diferentes, de manera que, tal como observó Wittmann en 1962, un aminoácido puede ser codificado por más de un codón.
En términos cuantitativos, se estima que 2 personas no relacionadas comparten más del 99% de sus secuencias de ADN, pero teniendo en cuenta los miles de millones de pares de bases que constituyen el genoma, las secuencias de ADN de las 2 personas no relacionadas pueden incluso variar en más de 20 millones de bases12,13. Algunos de estos cambios de base pueden resultar cruciales para incrementar el riesgo de enfermedad, tal como sucede en las denominadas enfermedades monogénicas. En ellas se observa una elevada penetrancia, y un solo cambio de base en un lugar del ADN puede dar una enfermedad bien caracterizada. Existen varios tipos bien documentados de fenotipos relacionados con enfermedades cardiovasculares con sustrato genético monogénico, entre las que se encontrarían la hipercolesterolemia familiar monocigótica por una o varias mutaciones en el gen del receptor de las lipoproteínas de baja densidad25. También predomina un componente monogénico en ciertas cardiopatías, como algunas congénitas y otras26. A pesar de la gran penetrancia de las enfermedades monogénicas, su frecuencia en la población es muy baja. Lo habitual es que las principales enfermedades cardiovasculares y sus fenotipos intermedios respondan a una herencia poligénica en la que concurren varios tipos de polimorfismos, y cada uno de ellos contribuye con un pequeño efecto que aumenta ligeramente el riesgo. En la tabla se presenta un glosario de términos clave relacionados con la genética y la epigenética cuyo significado es necesario conocer para comprender mejor los conceptos que se tratan en este artículo.
Glosario de términos en genética y epigenética
| Término | Descripción |
|---|---|
| ADN | El nombre químico de la molécula que lleva las instrucciones genéticas. Consiste en 2 hebras que se enrollan una alrededor de la otra para formar una doble hélice. Unida a cada azúcar hay 1 de 4 bases: adenina (A), citosina (C), guanina (G) y timina (T). Las 2 hebras se mantienen unidas por enlaces entre las bases con una correspondencia determinada |
| ARN | Molécula formada por un polirribonucleótido de longitud variable que contiene uracilo en vez de timina. Hay 3 tipos: ARN mensajero (ARNm), ARN ribosomal (ARNr) y ARN transferente (ARNt) |
| Alelo | Cada una de las versiones de un polimorfismo o gen. Un individuo típicamente hereda 2 alelos para cada polimorfismo, 1 de cada padre. Si los 2 alelos son iguales, el individuo es homocigótico para ese polimorfismo. Si son diferentes, es heterocigoto |
| Autosoma | Uno de los cromosomas no sexuales |
| Cromatina | Material formado por ácidos nucleicos y proteínas que se observa en el núcleo de la célula en interfase |
| Cromosoma | Paquete organizado de ADN que se encuentra en el núcleo de la célula. Cada organismo tiene un número de cromosomas diferente. Los humanos tienen 23 pares de cromosomas |
| Codón | Una secuencia de 3 bases de ADN o ARN que especifica 1 solo aminoácido en la traducción |
| Epigenético | Mecanismo de regulación de la expresión (transcripción y traducción) de genes que no depende de cambios en las bases del ADN, sino que opera a un nivel superior |
| EWA | Siglas en inglés de estudio de epigenoma completo. Se refiere a los estudios que analizan la metilación de epigenoma completo |
| Exón | Zona codificante de un gen |
| Fenotipo | Característica o rasgo observable de un individuo, fruto de la interacción entre su genotipo y el ambiente en que este se expresa. Se distinguen los fenotipos finales de enfermedad cardiovascular (infarto, ictus, etc.) y los fenotipos intermedios (hipertensión, dislipemias, etc.) |
| Gen | Unidad de herencia que ocupa una posición concreta en el genoma (locus) y tiene una estructura determinada |
| Genoma | Conjunto cromosómico básico que contiene toda la información genética del individuo |
| Genotipo | Suma de los alelos de un individuo para una posición determinada |
| GRS | Puntuación de riesgo genético. Puede ser ponderada o no ponderara en función de si valora el efecto de cada polimorfismo o solo los considera de manera aditiva |
| GWAS | Estudio de asociación de genoma completo |
| Histona | Proteínas pequeñas de carácter básico, ricas en lisina y arginina, que se unen al ADN en la cromatina |
| Locus | Lugar que un gen ocupa en el genoma |
| Metilación | Adición de restos metilo (-CH3) al ADN, en forma de bases metiladas |
| MicroARN | Fragmento muy pequeño de ARN no codificante y con importante función reguladora |
| Mutación | Cualquier cambio de base introducido en la secuencia de ADN. A veces se utiliza específicamente para indicar que la frecuencia alélica de la variación genética es muy baja (< 1%) |
| Nucleótido | Molécula constituida por una base nitrogenada, una pentosa y un grupo de ácido fosfórico. Unidad básica de la que se compone un ácido nucleico |
| Telómero | Extremo final de los cromosomas. En humanos, el ADN de los telómeros está compuesto por repeticiones en tándem de la secuencia TTAGGG |
En los últimos años, el desarrollo tecnológico para la determinación de las variaciones en el ADN ha sido espectacular, y actualmente se puede tener información muy precisa, rápida y económica sobre la presencia de determinadas variantes genéticas en el genoma de un individuo. El coste económico y el tiempo dependen del número de variantes genéticas que se quiera analizar. El primer paso en este proceso de análisis genómico comienza por la extracción de ADN. Para analizar ADN genómico, se puede utilizar cualquier muestra biológica de células nucleadas. Normalmente se utilizan leucocitos extraídos de sangre venosa periférica. El ADN extraído de esta muestra proporciona una concentración y una calidad adecuadas para la mayoría de los posteriores análisis genéticos. Como alternativa no invasiva a la obtención de muestras biológicas, se puede utilizar saliva, pero la concentración y la calidad del ADN extraído de ella puede no resultar suficiente para análisis genéticos masivos, por lo que en cada caso el investigador debe valorar los pros y contras de cada opción.
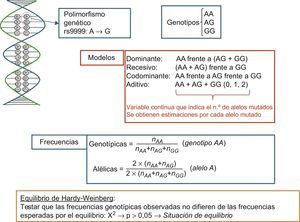
Si solamente se quiere analizar un polimorfismo o unos pocos polimorfismos en genes candidatos, la técnica utilizada es sobre todo la determinación de SNP basada en sondas fluorescentes; esta técnica reemplaza a la antigua basada en la utilización de enzimas de restricción y geles de agarosa, conocida como polimorfismos de longitud en los fragmentos de restricción. A la hora de denominar los polimorfismos, se ha adoptado una sistematización para identificar inequívocamente cada variante genética. Esta sistematización se basa en denominar el polimorfismos con un número de serie precedido por las letras rs («reference SNP»)14. Aquí sería bueno aclarar la diferencia entre los conceptos de mutación y de polimorfismo. Se trata de una diferenciación académica, ya que frecuentemente se utilizan como sinónimos. Ambas se refieren a cambios de base en el ADN, y la diferencia radica en su frecuencia. El término mutación se refiere a variantes muy poco comunes (con una frecuencia alélica del alelo menos frecuente < 1%), mientras que polimorfismo se refiere a las variaciones con frecuencias alélicas > 1%. En la figura 1 se presenta la situación de una variante en el ADN consistente en un cambio A>G (rs9999). Este cambio ocasiona 2 alelos (alelo A y alelo G). Estos alelos darán lugar a los 3 genotipos correspondientes, y a la hora de estudiar su asociación con los fenotipos de enfermedad, se puede utilizar distintos modelos de herencia (dominante, recesivo, codominante y aditivo). También se indica en la figura cómo se calculan las frecuencias alélicas y las frecuencias genotípicas, así como el test del equilibrio de Hardy-Weinberg.

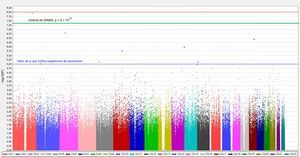
Para el estudio de un mayor número de polimorfismos, se usan distintos tipos de chips que permiten una genotipificación de más alta densidad. Entre estos chips se encuentran los denominados de «genoma completo». Ello hace referencia a que incluyen polimorfismos distribuidos por todos los cromosomas. Los primeros chips de genotipificación de genoma completo incluían la determinación de 10.000 SNP, también denominados de 10K (utilizando la letra K para designar los millares). Posteriormente se fue incrementando su densidad. Así por ejemplo, en los primeros estudios de asociación de genoma completo (genome-wide association studies [GWAS]) que se realizaron en el estudio de Framingham, publicados en 2007, se utilizaron chips de genotipificación de 100K27. Posteriormente se ha ido incrementando la densidad de estos chips, y actualmente incluyen densidades de genotipificación de más de 1 millón de SNP por individuo. En los análisis estadísticos de asociación entre la genotipificación de genoma completo y el fenotipo cardiovascular de interés28 se utilizan frecuentemente los gráficos denominados Manhattan plot, por analogía con los rascacielos de la isla de Manhattan en Nueva York. En la figura 2 se representa un Manhattan plot correspondiente a un GWAS de genotipificación densa con un array de 1.000K y un fenotipo intermedio cardiovascular. En el eje vertical se representa el valor del –log en base 10 de la p de asociación entre cada SNP y el fenotipo de interés, mientras que en el eje horizontal se representa la posición que ocupa cada SNP en el cromosoma. Cada punto de la gráfica es un SNP, de manera que solo se visualizan como puntos los SNP que tienen valores de p muy bajos, mientras que los demás SNP quedan como marcas compactas en la parte inferior de la gráfica. Cuanto más alto queda un SNP, más asociado se encuentra al fenotipo cardiovascular (su valor de la p de asociación es más pequeño). Para considerar estadísticamente significativa una asociación, no se utiliza el valor nominal de p < 0,05, sino que se corrige dicho valor por el número de comparaciones realizadas para minimizar los falsos positivos. El valor comúnmente aceptado como umbral para considerar estadísticamente significativa una asociación en GWAS es p < 5 × 10–8; este valor de p tiene su equivalente en el –log con un valor de 7,25. Algunos autores utilizan también el valor de p < 10–8, que tendría su equivalente en el –log en un valor de 8. Cuando se realiza un GWAS con fines exploratorios para confirmar posteriormente los resultados en otra población, por ejemplo, se establecen valores de p a partir de los cuales se considera que los resultados son sugerentes de asociación que confirmar posteriormente. Este valor es p < 10–5. Actualmente se han realizado centenares de GWAS con fenotipos intermedios y finales de enfermedad cardiovascular y se han identificado los principales genes y sus correspondientes SNP asociados con cada uno de ellos. Dado que una enumeración detallada de esos polimorfismos está fuera de los objetivos de esta revisión, se aconseja consultar las publicaciones originales o sus revisiones sintéticas6,28.

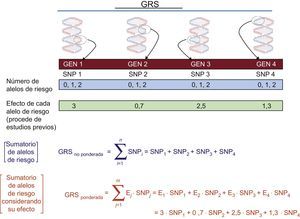
Los GWAS nos permiten conocer los principales SNP asociados con el fenotipo de interés por separado. Para conocer su contribución conjunta, se utilizan las denominadas puntuaciones de riesgo genético (genetic risk scores [GRS]). En la figura 3 se presenta el cálculo de las GRS en sus 2 principales modalidades: a) no ponderadas, y b) ponderadas. Múltiples estudios han analizado y cuantificado la influencia de varios GRS asociados con distintos fenotipos cardiovasculares29. Se puede encontrar en otras revisones30 un mayor detalle del cálculo de las GRS y sus ventajas e inconvenientes.

Paralelamente, se dispone también de la gran mejora tecnológica en la secuenciación directa que ahora se denomina next generation sequencing (NGS), ya que, en lugar de basarse en las técnicas aplicadas en el Proyecto Genoma Humano (método de secuenciación de electroforesis capilar tradicional de Sanger, considerada la primera generación), las técnicas de NGS proporcionan un mayor rendimiento de datos a menor coste. La NGS tiene 3 mejoras importantes31: a) no requiere un procedimiento de clonación bacteriana y prepara las bibliotecas para la secuenciación en un sistema libre de células; b) se procesan millones de reacciones de secuenciación en paralelo y al mismo tiempo, y c) la detección de bases se realiza cíclicamente y en paralelo. Todo ello aumenta mucho la fiabilidad y disminuye el coste. Actualmente existen varios proyectos que tienen como objetivo secuenciar el genoma de miles de individuos y mapearlos; a facilitar este objetivo está contribuyendo la tecnología con el HiSeq X, secuenciador de alto rendimiento de Illumina. El HiSeq X produce 1,8 Tb de secuencia por ejecución en 3 días y está especialmente diseñado para la secuenciación del genoma completo, que requiere un rendimiento muy alto y secuenciación multiparalela al mismo tiempo31. El objetivo de secuenciar completamente un genoma humano por 1.000 dólares está a la vista, y el coste puede ser incluso inferior con las denominadas tecnologías de tercera generación31. Actualmente, la secuenciación de exomas ya está bastante extendida en los estudios de genética cardiovascular32 y paulatinamente lo estará la secuenciación del genoma completo.
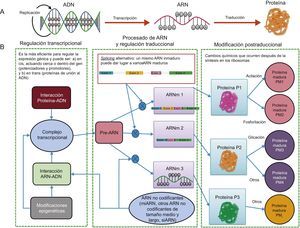
EPIGENÓMICAA pesar de los grandes avances realizados en la secuenciación del genoma, el conocimiento de las variantes genéticas en el ADN no es suficiente para predecir el riesgo de enfermedad, ya que existen otros elementos reguladores más dinámicos, denominados epigenoma, que a su vez tienen capacidad para regular la expresión de las secuencias de ADN. Por ello hoy resulta imprescindible estudiar conjuntamente el genoma y el epigenoma para un mejor conocimiento de las bases moleculares en los fenotipos de riesgo cardiovascular. La existencia de regulaciones epigenéticas ayuda a comprender por qué no se cumple el dogma fundamental de la biología (figura 4). En el panel A se representa el proceso clásico en el que un ADN se transcribe a un ARN y este resulta en una proteína, mientras que en el panel B se representa la situación más compleja reconocida actualmente, mediante la cual un solo ADN puede dar lugar a distintas proteínas debido a la influencia de distintos procesos epigenéticos reguladores que van más allá de las mutaciones en el ADN13. La epigenómica es el estudio de los elementos funcionales clave que regulan la expresión génica en una célula. A diferencia del genoma, que es el mismo en todas las células somáticas, el epigenoma es específico de cada tipo celular, lo cual añade más complejidad al estudio y hace muy relevante el origen de la muestra que se ha tomado para el análisis. Habrá diferencias en los resultados del análisis del epigenoma si se analizan leucocitos o tejido adiposo, por ejemplo, aunque sean de una misma persona20. Para analizar el epigenoma de los distintos tipos celulares, varios consorcios, reunidos bajo el paraguas del Consorcio Internacional del Epigenoma Humano, han asumido el reto de descifrar cientos de epigenomas humanos sanos y enfermos, específicos de cada tipo celular, y difundir los resultados en varias publicaciones33. Existen muchos tipos de modificaciones epigenéticas, las más estudiadas son las metilaciones y las regulaciones por ARN no codificantes, entre las que se encontrarían los microARN, pero también otros tipos de ARN. Las modificaciones de histonas, otro tipo de regulación epigenética, tienen un mayor nivel de complejidad y todavía están poco estudiadas en ensayos epidemiológicos en humanos. El gran interés de profundizar en el conocimiento de los reguladores epigenéticos radica en que son dinámicos. A diferencia de los SNP en el ADN, las marcas epigenéticas pueden modificarse, y el conocimiento de los factores que intervienen en esta modificación puede ser crucial para la prevención y/o el tratamiento de las enfermedades cardiovasculares.

Transcripción y traducción. A: según dogma central de la biología, en el que un gen daba lugar a una sola proteína. B: visión más moderna, que incluye varios elementos reguladores adicionales que hacen que un mismo gen pueda dar lugar a varias proteínas. ARNm: ARN mensajero; miARN: microARN; PM: proteína madura; Pre–ARN: ARN precursor; siARN: ARN de silenciamiento.
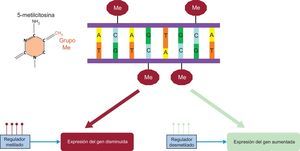
Esta modificación epigenética en el ADN se produce por la adición enzimática de un grupo metilo al carbono 5 de la citosina (figura 5). La mayoría de las 5-metilcitosinas están presentes en los dinucleótidos de citosina-fosfato-guanina (CpG). La metilación del ADN se realiza por las ADN metiltransferasas. Estás se clasifican en 2 grupos: ADN metiltransferasas de mantenimiento (DNMT1), que son la que mantienen los patrones de metilación, y de novo (DNMT3A y DNMT3B), que son las que realizan nuevas metilaciones. También existen desmetilasas que se encargarían del proceso inverso de eliminación de los grupos metilo, pero estos mecanismos son menos conocidos. Un mayor detalle sobre todos estos mecanismos de metilación y desmetilación se puede encontrar en revisiones temáticas específicas20,34,35. Los dinucleótidos CpG no están distribuidos de manera uniforme en todo el genoma y se concentran en algunas zonas. Estas zonas ricas en dicho dinucleótidos (más del 60%) se denominan islas CpG. Estas islas suelen concentrarse entre el promotor y la zona del inicio de la transcripción. La regulación epigenética por metilación es compleja, pero en general la metilación en elementos reguladores de los genes, tales como promotores, potenciadores, aislantes y represores, suprime su función35 (figura 5). Hay varias formas de analizar la metilación de un gen concreto o por todo el genoma en general. Para ello, al igual que lo comentado sobre la genotipificación del genoma completo, para el estudio del epigenoma completo (EWA) (metilación), hay chips específicos que analizan las zonas más importantes del genoma. Inicialmente se utilizó el chip de metilación de 450K de Illumina, y recientemente se ha sustituido por un chip de mayor cobertura, también de Illumina, que cubre 850K36. Para analizar la asociación, también se utilizan los gráficos tipo Mahattan plot, esta vez valorando los valores de p de la asociación entre metilación y el fenotipo específico37. Aunque se han publicado varios estudios de EWA, la concordancia de los resultados entre ellos todavía es baja. Considerando otros tipos de metilación, una revisión analizó los resultados de 31 artículos centrados en la metilación y los lípidos plasmáticos que incluyeron en total a 8.027 participantes38. En general, no se observaron asociaciones firmes entre la metilación general del ADN y los lípidos plasmáticos. En cuanto a la metilación de genes específicos, encontraron resultados que se repetían con los genes ABCG1, CPT1A, TNNT1, MIR33B, SREBF1 y TNIP38, por lo que hay que seguir profundizando en estas relaciones y sus moduladores. Se sabe que la dieta y el consumo de tabaco son factores importantes que influyen en la metilación del ADN39, por lo que también hay que valorar bien su influencia en los estudios de EWA.

Recientemente también está despertando gran interés la regulación por hidroximetilación del ADN40. Este proceso se lleva a cabo por enzimas TET (ten-eleven translocation methylcytosine dioxygenase), que reconocen ciertas citosinas anteriormente metiladas y las oxidan, lo que causa el paso de la 5-metilcitosina a 5-hidroximetilcitosina. Parecería que los genes hidroximetilados se asocian a un aumento de la transcripción. Al ser un campo más nuevo, son necesarios muchos más estudios para valorar su influencia en el riesgo cardiovascular.
Regulación por proteínas de unión a metilcitosina-fosfato-guaninaEste mecanismo involucra proteínas o complejos proteínicos que se unen específicamente a lugares CpG metilados y bloquean indirectamente la unión de los factores de transcripción al limitar su acceso a los elementos reguladores35. Estas proteínas contienen dominios conservados de unión a ADN metilado (denominados en inglés methyl binding domain [MBD]). La primera proteína identificada fue la MeCP2, y posteriormente se caracterizaron otras proteínas, entre ellas: MBD1, MBD2 y MBD3, implicadas en la represión transcripcional, al igual que MeCP2. Otras proteínas MBD tienen distintas funciones. En general la regulación es compleja35.
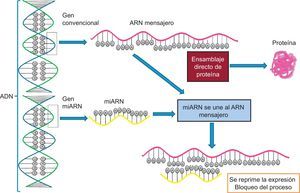
Regulación por ARN no codificantes. MicroARN y otrosRecientemente se ha identificado un gran número de ARN no codificantes. Durante años se desconoció su función, pero actualmente se está avanzando en la comprensión de su funcionalidad41. Estos ARN no codifican proteínas y tienen una importante función reguladora de múltiples procesos. Se clasifican en ARN no codificantes cortos (menos de 200 pb) y ARN no codificantes largos (más de 200 pb)19. Los microARN son los más pequeños (∼20-25 pb) y los más estudiados. Los microARN se codifican en el genoma, bien sea en los intrones dentro de otro gen codificante, o en los espacios intergénicos. Se transcriben como un transcripto primario bicatenario (pri-miR) de mayor tamaño por la ARN polimerasa II. Posteriormente, la enzima nuclear Drosha (también conocida como ribonucleasa III) y Pasha convierten este precursor en un precursor de microARN bicatenario de ∼70 nulcleótidos (pre-miR), y un mecanismo que implica la proteína exportina lo transportan al citoplasma. Finalmente, la enzima Dicer procesa este precursor en un microARN de doble cadena de 22 nucleótidos. Este dúplex se desenrolla, y 1 de las 2 hebras se incorpora en el RISC (complejo silenciador inducido por ARN), que comprende la proteína argonauta y otras42. Los microARN incorporados en el RISC son capaces de unirse a la región 3’ UTR de los ARN mensajeros objetivo y causar un bloqueo de la traducción. La otra hebra de microARN se degrada. En la figura 6 se esquematiza el proceso de unión del microARN a su ARN mensajero diana que bloquea la producción de proteína, en comparación con el proceso sin el bloqueo por el microARN. Existen múltiples microARN implicados en la regulación de los distintos fenotipos cardiovasculares. Se puede analizar estos microARN en tejidos específicos y relacionar su expresión en ellos con ciertas características fenotípicas, como por ejemplo la relación entre la expresión de los microARN miR-1, miR-133 y miR-208 y el desarrollo de hipertrofia ventricular e insuficiencia cardiaca. Otras veces se puede encontrar y analizar los microARN en el plasma circulante, y sus concentraciones pueden ser indicadoras de varias enfermedades o procesos. Por ejemplo, los miR-1, miR-133a, miR-133b y miR-499-5p están elevados en plasma tras un infarto agudo de miocardio42. Este campo emergente de investigación, a pesar de su gran profusión, todavía necesita una mejor estandarización de técnicas y procesos, ya que está sujeto a una variación muy dinámica. Tras esta estandarización, se hallarán sin duda múltiples aplicaciones en este apasionante mundo de regulación epigenómica.
Regulación por modificación de histonas
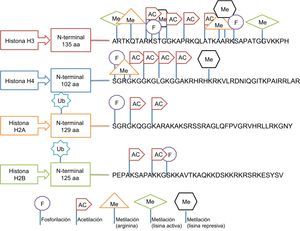
Este tipo de regulación epigenética es más complejo y está menos analizado en los estudios epidemiológicos en humanos. Las principales modificaciones postraduccionales que pueden tener lugar en las histonas incluyen, entre otras: acetilación, fosforilación, metilación o ubiquitinización, e influyen en el estado de compactación de la cromatina20. Las histonas tienen un dominio carboxilo terminal globular y una cola aminoterminal, que es donde tienen lugar las modificaciones. Pero estas modificaciones no ocurren en cualquier sitio, sino que responden a unos códigos de regulación muy estructurados. Así, por ejemplo, las metilaciones ocurren en los residuos de lisina (abreviada como K) y arginina (R); las acetilaciones, en residuos de lisina; la ubiquitinación, en lisinas; la fosforilación, en serinas (S) y treoninas (T), etc. También influye en el resultado el tipo de histona en que se produce la modificación35. En la figura 7 se presenta gráficamente este proceso selectivo de modificación de histonas. A pesar de las dificultades, se han realizado algunos estudios que han reportado que la HDAC3 (histona deacetilasa 3) desempeña un papel crítico en la función endotelial, mientras que la HDAC7 (histona deacetilasa 7) es relevante en la función de las células musculares lisas vasculares35. Asimismo, se ha reportado que las enzimas de las histonas están más infraexpresadas que sobrexpresadas cuando acontecen enfermedades metabólicas43. Sin embargo, todavía hay que estandarizar diferencias de expresión según los tejidos y otros reguladores complejos que también intervienen en los procesos35.
INTEGRACIÓN DE GENÓMICA Y EPIGENÓMICA. INTERACCIONES GEN-AMBIENTE.")
Aunque se han presentado por separado los conceptos genómicos y epigenómicos, la realidad es que se dan en interacción. Se sabe que las metilaciones a su vez dependen de algunos polimorfismos genéticos que haya en la secuencia44. Del mismo modo, la unión de un microARN al ARN mensajero diana no será la misma si hay una determinada secuencia en bases o si tiene lugar un polimorfismo genético44. En este sentido, se puede indicar como ejemplo los interesantes resultados que obtuvimos analizando el polimorfismo en un lugar de unión de un microARN (en este caso el miR410) en el gen de la lipoproteinlipasa en las concentraciones plasmáticas de triglicéridos y su posterior modulación por la dieta mediterránea para determinar el riesgo de ictus45. Todo ello lleva a la necesidad de plantear estudios integrados, no solo de ómicas, sino también de factores ambientales como la dieta y otras variables del estilo de vida, para conocer mejor las bases moleculares y su regulación en el riesgo cardiovascular. Para el estudio de las interacciones gen-ambiente, hasta ahora había preponderado la visión de interacción estadística, en lugar de prestar atención también a las interacciones biológicas (véase una revisión46 para entender mejor la diferencia entre ellas). Sin embargo, la publicación de una importante interacción biológica, no estadísticamente significativa, entre una GRS de riesgo cardiovascular y el estilo de vida, que determina la incidencia de enfermedad cardiovascular47, ha contribuido a reforzar el concepto de interacción gen-ambiente biológica, entendida como la existencia de un factor ambiental que puede modificar una susceptibilidad genética, y en ese sentido se plantean nuevos estudios desde esta perspectiva.
FINANCIACIÓNEste trabajo ha contado con el apoyo del Ministerio de Salud (Instituto de Salud Carlos III) y del Ministerio de Economía y Competitividad (CIBER 06/03, CNIC-06/2007, RTIC G03/140, SAF2016-80532-R, AES_FIS_2016), y la Fundació La Marató de TV3 (538/U/2016).
CONFLICTO DE INTERESESNinguno.