
La miocardiopatía arritmogénica del ventrículo derecho (MAVD), descrita por primera vez en 1977 en pacientes a los que se había practicado una ablación quirúrgica por una taquicardia del tracto de salida del ventrículo derecho (TSVD)1, es una enfermedad del músculo cardiaco caracterizada por pérdida de miocardiocitos y su sustitución por tejido fibroso o fibroadiposo, que puede dar lugar a arritmias, muerte súbita cardiaca e insuficiencia cardiaca. La enfermedad se hereda con frecuencia en forma de rasgo autosómico dominante causado por mutaciones en genes que codifican proteínas desmosómicas2, 3, 4, 5, 6, 7, 8 y no desmosómicas9, 10, 11, 12, 13, 14. El análisis de la expresión de la enfermedad en los portadores de mutaciones y sus familiares ha puesto de manifiesto que la MAVD es una enfermedad compleja, lo cual dificulta su definición clínica y el diagnóstico. En 1994, el Grupo de Trabajo en Enfermedades del Pericardio y el Miocardio de la Sociedad Europea de Cardiología y el Scientific Council on Cardiomyopathies de la International Society and Federation of Cardiology publicaron unos criterios diagnósticos15. Dichos criterios se basaban en un sistema de puntuación que tenía en cuenta las anomalías morfológicas y funcionales del ventrículo derecho (VD), las características del electrocardiograma (ECG), los resultados histopatológicos, las arritmias ventriculares y los antecedentes familiares. Los criterios fueron modificados en 201016 para mejorar su sensibilidad en el diagnóstico precoz y en la enfermedad de carácter familiar. La presente revisión se centra en los nuevos criterios de la MAVD.
Perspectiva históricaEl concepto de una miocardiopatía específica del VD se propuso por primera vez en una descripción de 6 pacientes con taquicardia ventricular (TV) sostenida y agrandamiento del VD que se publicó en 19771. En 1982, se utilizó por primera vez el término «displasia ventricular derecha arritmógena» en la presentación de una serie de 24 casos de pacientes con un patrón de bloqueo de rama izquierda del haz (BRIH), TV, anomalías del movimiento de la pared del VD y sustitución del miocardio del VD por tejido adiposo y fibroso17. Sin embargo, la displasia ventricular derecha arritmógena (o MAVD, como se la denominó más tarde) no se reconoció formalmente como una entidad diferenciada hasta 1994, tras la publicación de los criterios diagnósticos de la Task Force de la Organización Mundial de la Salud/International Society and Federation of Cardiology15. Esos criterios se basaban en la identificación de anomalías estructurales, sustitución fibroadiposa del miocardio del VD, alteraciones características en el ECG, arritmias de origen en el VD y evidencia de enfermedad de carácter familiar.
Las primeras mutaciones génicas ligadas a la enfermedad se identificaron en una variante sindrómica de la MAVD, de carácter recesivo, denominada enfermedad de Naxos. Se observó que la causa de esta forma especialmente maligna del trastorno era una deleción de dos pares de bases en el gen que codifica la placoglobina2, un componente importante de las uniones intercelulares. Esto abrió el camino a la identificación de mutaciones causantes de la enfermedad en otros genes que codifican proteínas desmosómicas en las formas autosómicas dominantes más frecuentes de la MAVD3, 4, 5, 6, 7, 8. Posteriormente se han identificado fenotipos clínicos e histológicos muy similares en pacientes con mutaciones en genes no desmosómicos, como los de titina, desmina y lamina A/C9, 10, 11, 12, 13, 14.
EpidemiologíaLa prevalencia de la MAVD en la población general es difícil de estimar debido a las dificultades que comporta el diagnóstico. Los estudios realizados en Europa indican una prevalencia de entre 0,6 y 4,4‰, pero es posible que estas cifras estén sesgadas por las diferencias geográficas existentes en la disponibilidad de expertos clínicos y anatomopatológicos. La MVDA se notifica como causa de muerte súbita cardiaca en un 11-27% de los casos de pacientes de edad ≤ 35 años18, 19, 20. La prevalencia de muerte súbita en una cohorte italiana fue mayor en deportistas (22,4%) que en no deportistas (8,2%)18.
Criterios de 2010El carácter inespecífico de la mayor parte de los signos clínicos y la ausencia de una prueba diagnóstica única hacen que el diagnóstico de la MAVD resulte siempre difícil. Este grado de dificultad se reflejó en primer lugar en la guía de la International Task Force de 1994, que propuso unos criterios estandarizados para el diagnóstico basados en la identificación de características estructurales, histológicas, electrocardiográficas, arrítmicas y familiares, que luego se subdividieron en criterios mayores y menores según la especificidad percibida15. Se consideró diagnóstica la detección de dos criterios mayores, un criterio mayor junto con dos menores o cuatro criterios menores de diferentes categorías. La experiencia clínica posterior con el empleo de estas guías indicó que, aunque los criterios son muy específicos, carecen de sensibilidad para la enfermedad en una fase temprana. También, muchos de los parámetros eran subjetivos y sin base en evidencia. Por este motivo, en 2010 se propusieron nuevos criterios16. El esquema básico es el mismo, pero la terminología diagnóstica para los nuevos criterios se ha modificado de la siguiente forma:
• Diagnóstico definitivo: dos criterios mayores o un criterio mayor y dos menores, o cuatro criterios menores de categorías diagnósticas diferentes.
• Diagnóstico limítrofe: un criterio mayor y un criterio menor, o tres criterios menores de categorías diagnósticas diferentes.
• Diagnóstico posible: un criterio mayor o dos criterios menores de categorías diagnósticas diferentes.
En el apartado siguiente se comentan los cambios más relevantes introducidos en dominios diagnósticos específicos en el nuevo esquema.
Diagnóstico por la imagenAlgunas de las características estructurales y funcionales del VD plantean importantes dificultades para el diagnóstico por imagen ecocardiográfica (Figura 1). Concretamente, se debe a su posición próxima a la pared anterior del tórax y a su forma, su orientación y su geometría complejas y su pared delgada. En comparación con la ecocardiografía, la resonancia magnética cardiaca (RMC) aporta varias ventajas que permiten superar algunas de estas dificultades. Entre ellas se encuentra la visualización tridimensional del corazón y su relación con las estructuras torácicas, la ausencia de supuestos geométricos en la cuantificación de los volúmenes del VD y el ventrículo izquierdo (VI), una resolución espacial excelente y la caracterización del tejido. Sin embargo, también tiene limitaciones, puesto que la pared del VD es muy delgada y la imagen puede verse afectada por un efecto de volumen parcial. Además, puede resultar difícil diferenciar entre la grasa pericárdica y la infiltración grasa del VD. Por último, el desconocimiento sobre las variaciones normales de la morfología del VD da lugar a una gran variabilidad interobservador e intraobservador en la presentación de los resultados.

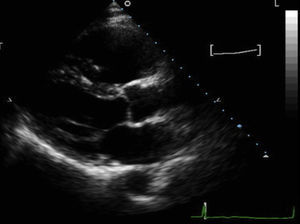
Figura 1. Proyección de eje largo paraesternal de un paciente con una miocardiopatía arritmogénica ventricular derecha, que muestra una intensa dilatación ventricular derecha (tracto de salida ventricular derecho 4,7 cm); el paciente presentaba una disfunción sistólica ventricular derecha grave y una pared del tracto de salida ventricular derecho acinética.
El papel de la RMC en el diagnóstico de la MAVD se ha reconocido en los nuevos criterios de la Task Force. Mientras que los criterios de 1994 se basaban en la evaluación cualitativa del tamaño y la función del VD (p. ej., «intensas dilatación y reducción de la fracción de eyección del VD» o «leves dilatación general y/o reducción de la fracción de eyección del VD»), los nuevos criterios introducen valores de corte precisos para la evaluación del VD. Además, se ha excluido de los criterios la presencia de anomalías leves en la motilidad regional de la pared (hipocinesia), ya que tiene una especificidad muy baja.
Uno de los principales cambios que se plantean en los nuevos criterios de diagnóstico por imagen es que reconoce que la afección del VI es parte del espectro clínico de la enfermedad. Los criterios de 1994 exigían escaso o nulo deterioro del VI para preservar una especificidad elevada y evitar la inclusión de pacientes con miocardiopatía dilatada. En realidad, varios estudios han puesto de relieve que la afección del VI es frecuente con la progresión de la enfermedad y puede constituir un indicador pronóstico21. Recientemente se han descrito formas con afección predominante del VI22, aunque en ese caso la terminología es muy confusa y algo arbitraria. En resumen, la presencia de dilatación o disfunción del VI ha dejado de ser un criterio de exclusión para el diagnóstico.
Debe señalarse que, si bien la RMC puede usarse para evaluar la fibrosis y la infiltración adiposa, su utilidad en la pared delgada del VD continúa siendo objeto de controversia, y la caracterización del tejido no forma parte de los criterios diagnósticos. Por otro lado, la captación tardía de contraste con gadolinio y, de manera menos uniforme, una infiltración adiposa del miocardio, en especial en el VI, pueden ser útiles para identificar a los individuos afectados.
Caracterización del tejidoEl patrón de referencia para el diagnóstico de la MAVD en los criterios de 1994 era la demostración de una sustitución fibroadiposa transmural del miocardio ventricular derecho en el tejido obtenido en la autopsia, la cirugía o una biopsia endomiocárdica23. En este contexto, la biopsia endomiocárdica tiene ciertas limitaciones intrínsecas, entre ellas la no menor de que no proporciona una muestra transmural, que es importante porque la enfermedad suele evolucionar desde el epicardio hacia el endocardio. Además, las muestras de biopsia endomiocárdica suelen obtenerse del tabique interventricular, que se ve afectado con menor frecuencia que otras regiones del VD. No obstante, se han propuesto parámetros histomorfométricos para el diagnóstico de la MAVD, concretamente, una atrofia miocárdica con miocitos residuales < 45% del área de corte transversal, tejido fibroso > 40% y tejido adiposo > 3%. Se ha descrito que estos criterios tienen una sensibilidad del 67% y una especificidad del 92% para el diagnóstico23.
En un estudio de evaluación cuantitativa de la biopsia endomiocárdica en la MAVD en corazones explantados24, se observó una considerable variabilidad de las anomalías histológicas entre diferentes lugares de obtención de las muestras dentro del VD; el área anteroapical resultó ser la más informativa. Es importante señalar que no se identificó ningún valor de corte diagnóstico útil para el tejido adiposo en el área anteroapical y del TSVD. Los parámetros con una especificidad del 95% fueron los siguientes: miocardio residual < 59%, fibrosis > 31% y grasa > 22%. El valor de corte para la grasa dependía de la edad y la obesidad y, al excluir a los pacientes ancianos y a los obesos, se estableció un valor de corte inferior para la grasa (> 9%). A la vista de estos resultados, en la modificación de 2010 se han propuesto nuevos criterios histológicos específicos mayores y menores (Tabla).
Criterios de 2010 de la Task Force para el diagnóstico de la miocardiopatía arritmogénica ventricular derecha
| I. Disfunción y alteraciones estructurales globales o regionales | |
| Mayores | En el ecocardiograma bidimensional: acinesia, discinesia o aneurisma regionales del VD y 1 de las siguientes (en telediástole):• PELP TSVD ≥ 32 mm (corregido por tamaño corporal [PELP/ASC] ≥ 19 mm/m2)• PECP TSVD ≥ 36 mm (corregido por tamaño corporal [PECP/ASC] ≥ 21 mm/m2)• o cambio del área fraccional ≥ 33% |
| En la RM: acinesia o discinesia regionales del VD o contracción disincrónica del VD y 1 de las siguientes:• Cociente de volumen telediastólico del VD respecto a ASC ≥ 110 ml/m2 (varones) o ≥ 100 ml/m2 (mujeres)• O fracción de eyección del VD ≤ 40% | |
| En la angiografía del VD: acinesia, discinesia o aneurisma regionales del VD | |
| Menores | En el ecocardiograma bidimensional: acinesia o discinesia regionales del VD y 1 de las siguientes (en el periodo telediastólico):• PELP TSVD ≥ 29 a < 32 mm (corregido por tamaño corporal [PELP/ASC] ≥ 16 a < 19 mm/m2)• PECP TSVD ≥ 32 a < 36 mm (corregido por tamaño corporal [PECP/ASC] ≥ 18 a < 21 mm/m2)• O cambio del área fraccional > 33 a ≤ 40% |
| En la RM: acinesia o discinesia regionales del VD o contracción disincrónica del VD y 1 de las siguientes:• Cociente de volumen telediastólico del VD respecto a ASC ≥ 100 a < 110 ml/m2 (varones) o ≥ 90 a < 100 ml/m2 (mujeres)• O fracción de eyección del VD > 40 a ≤ 45% | |
| II. Caracterización del tejido de la pared | |
| Mayores | Miocitos residuales < 60% mediante análisis morfométrico (o < 50% si se han estimado), con sustitución fibrosa del miocardio de la pared libre del VD en al menos una muestra, con o sin sustitución adiposa del tejido en la biopsia endomiocárdica |
| Menores | Miocitos residuales del 60 al 75% mediante análisis morfométrico (o del 50 al 65% si se han estimado), con sustitución fibrosa del miocardio de la pared libre del VD en al menos una muestra, con o sin sustitución adiposa del tejido en la biopsia endomiocárdica |
| III. Anomalías de la repolarización | |
| Mayores | Ondas T invertidas en las derivaciones precordiales derechas (V1, V2 y V3) o más allá en individuos de edad > 14 años (en ausencia de BRDH con QRS ≥ 120 ms) |
| Menores | Ondas T invertidas en las derivaciones V1 y V2 en individuos de edad > 14 años (en ausencia de BRDH completo) o en V4, V5 o V6Ondas T invertidas en las derivaciones V1, V2, V3 y V4 en individuos de edad > 14 años en presencia de un BRDH completo |
| IV. Anomalías de despolarización/conducción | |
| Mayores | Onda épsilon (señales de baja amplitud reproducibles entre el final del complejo QRS y el inicio de la onda T) en las derivaciones precordiales derechas (V1 a V3) |
| Menores | Potenciales tardíos mediante SAECG en al menos uno de tres parámetros, en ausencia de una duración del QRS ≥ 110 ms en el ECG estándar: duración del QRS filtrado ≥ 114 ms; duración del QRS terminal < 40 μV (duración de señal de baja amplitud) ≥ 38 ms; raíz de la media cuadrados de los voltajes de los 40 ms terminales ≤ 20 μVDuración de la activación terminal del QRS ≥ 55 ms medida desde el mínimo de la onda S hasta el final del QRS, incluyendo R’, en V1, V2 o V3, en ausencia de BRDH completo |
| V. Arritmias | |
| Mayores | Taquicardia ventricular no sostenida o sostenida con morfología de BRIH con eje superior (QRS negativo o indeterminado en las derivaciones II, III, aVF; y positivo en aVL) |
| Menores | Taquicardia ventricular no sostenida o sostenida de configuración de TSVD, con morfología de BRIH con eje inferior (QRS positivo en las derivaciones II, III y aVF y negativo en aVL) o de eje desconocido> 500 extrasístoles ventriculares por 24 h (Holter) |
| VI. Antecedentes familiares | |
| Mayores | M/DAVD confirmada en un familiar de primer grado que cumpla los criterios actuales de la Task ForceM/DAVD confirmada anatomopatológicamente en la autopsia o la intervención quirúrgica en un familiar de primer gradoIdentificación de una mutación patogénica * clasificada como asociada o probablemente asociada a la M/DAVD en el paciente examinado |
| Menores | Antecedentes de M/DAVD en un familiar de primer grado en el que no es factible determinar si cumple los criterios actuales de la Task ForceMuerte súbita prematura (< 35 años de edad) debida a presunta M/DAVD en un familiar de primer gradoM/DAVD confirmada anatomopatológicamente o mediante los criterios actuales de la Task Force en un familiar de segundo grado |
ASC: área de superficie corporal; aVF: aumento de voltaje en derivación unipolar del pie izquierdo; aVL: aumento de voltaje en derivación unipolar de brazo izquierdo; BRDH: bloqueo de rama derecha del haz; BRIH: bloqueo de rama izquierda del haz; ECG: electrocardiograma; M/DAVD: miocardiopatía/displasia arritmogénica ventricular derecha; PECP: proyección de eje corto paraesternal; PELP: proyección de eje largo paraesternal; RM: resonancia magnética; SAECG: electrocardiograma de promediación de señal; TSVD: tracto de salida del ventrículo derecho; VD: ventrículo derecho.
* Una mutación patógena es una alteración del ADN asociada a la M/DAVD que altera o se prevé que alterará la proteína codificada, no se observa o es muy poco común en una población de control amplia sin M/DAVD y bien altera o se predice que alterará la estructura o la función de la proteína, bien presenta un ligamiento demostrado con el fenotipo de la enfermedad en un examen genealógico concluyente.Modificado de Marcus et al 16 con permiso del editor.
Recientemente se ha propuesto para el diagnóstico de la MVDA una nueva prueba inmunohistoquímica que determina la localización de la placoglobina en la biopsia endomiocárdica25. Sin embargo, la prueba no es específica para la MVDA, puesto que también es positiva en los pacientes con sarcoidosis o con miocarditis de células gigantes26.
Anomalías de la repolarizaciónEl ECG es clave en el diagnóstico de la MVDA (Figura 2) y las anomalías en él se han resaltado en los nuevos criterios modificados. Una de las características clave de la MAVD es la presencia de una inversión de la onda T en las derivaciones precordiales derechas, sin que haya un bloqueo de rama derecha del haz (BRDH) completo. La prevalencia de la inversión de la onda T en las derivaciones precordiales derechas en sujetos sanos de más de 14 años de edad ha sido de tan sólo un 4% en las mujeres y un 1% en los varones27. Por consiguiente, ahora este criterio se considera lo bastante específico para constituir una anomalía mayor, mientras que en los criterios de 1994 era de tipo menor. Además, se ha introducido como anomalía menor la presencia de una inversión de la onda T en V1-V2 (en individuos de más de 14 años de edad y en ausencia de un BRDH completo). La identificación de que puede darse una afección del VI o que esta puede ser incluso la característica principal de la MAVD se refleja también en los criterios de repolarización, que incluyen la presencia de inversión de la onda T en las derivaciones laterales, V4, V5 o V6 (que se observan de forma característica en casos de afección del VI) como criterio menor.

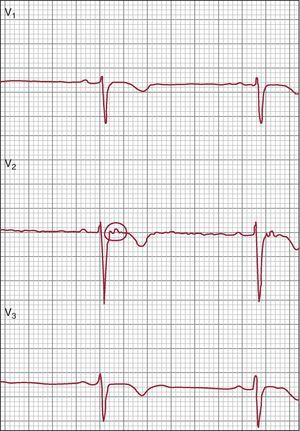
Figura 2. Electrocardiograma de un paciente con una miocardiopatía arritmogénica ventricular derecha, en el que se observa ritmo sinusal, mala progresión de la onda R e inversión de la onda T en las derivaciones precordiales derechas.
En un estudio de Jain et al28 se investigó sistemáticamente el valor de las características del ECG en la MAVD incluyendo a pacientes con BRDH incompleto o completo. En los pacientes con MAVD y BRDH completo, los dos únicos parámetros que diferían respecto a la población de control fueron la presencia de inversión de la onda T hasta el nivel de V4 (el 59 frente al 12%, respectivamente) y el cociente r’/s en V1 < 1 (el 88 frente al 14%). A la vista de estas observaciones, la presencia de inversión de la onda T en V1-V4 en sujetos de más de 14 años de edad con BRDH completo se considera un criterio menor.
Anomalías de despolarización/conducciónLas ondas épsilon (Figura 3) son potenciales eléctricos de baja amplitud que se producen al final del complejo QRS y al inicio del segmento ST y que se detectan en las derivaciones precordiales derechas. Su identificación puede verse facilitada por el empleo de un filtro de paso alto de 40 Hz, que aumenta la ganancia a 20 mV/mm y la velocidad de registro a 50 mm/s. Una modificación de la posición de las derivaciones de las extremidades (derivación del brazo derecho colocada en el manubrio esternal, derivación del brazo izquierdo colocada sobre el xifoides y derivación de la pierna izquierda colocada sobre una costilla entre las posiciones habituales de V4 y V5) puede mejorar la sensibilidad30. Se cree que las ondas épsilon corresponden a áreas de activación retardada del VD como consecuencia de la sustitución fibrosa o fibroadiposa del miocardio del VD, lo que se considera un criterio mayor.

Figura 3. Onda épsilon en V2 (círculo). Reproducido de Quarta et al 29 con permiso del editor.
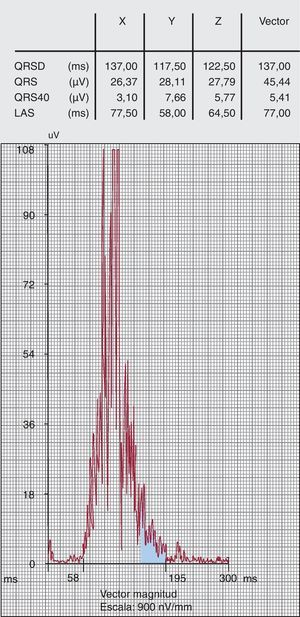
La presencia de potenciales tardíos detectados mediante el ECG de promediación de señal (Figura 4) indica áreas de conducción ventricular lenta, que pueden corresponder a un sustrato anatómico para arritmias ventriculares de reentrada. El ECG de promediación de señal se realiza con un filtro de 40-250 Hz para asegurar un nivel de ruido < 0,3 μV. Se analizan habitualmente los tres parámetros siguientes: duración del QRS filtrado, duración de la señal de baja amplitud (< 40 μV) del QRS terminal (LAS) y raíz cuadrada de la media de amplitudes al cuadrado de los últimos 40 ms de la señal de QRS (QRS40). En los criterios originales, se consideraba que había una presencia de potenciales tardíos si dos de estos parámetros eran anormales: concretamente, QRS filtrado ≥ 114 ms, LAS ≥ 38 ms y QRS40 ≤ 20 μV. Sin embargo, estos valores no estaban basados en la evidencia. La sensibilidad y la especificidad del ECG de promediación de señal se ha comparado en 69 pacientes con MVDA frente a 103 controles normales16: cada uno de los parámetros tuvo una sensibilidad del 58-60% y una especificidad del 94-96%, mientras que la presencia de dos parámetros mostró valores de sensibilidad (66%) y especificidad (95%) similares; el empleo de la presencia de cualquiera de los parámetros tuvo una sensibilidad del 74% y una especificidad del 92%. A la vista de estas observaciones en el nuevo algoritmo diagnóstico, la presencia de uno o varios parámetros (en ausencia de una duración del QRS ≥ 110 ms) se considera un criterio menor.

Figura 4. Electrocardiograma de promediación de señal positivo para potenciales tardíos. LAS: duración de la señal de baja amplitud (< 40 μV) del QRS terminal; QRS40: raíz cuadrada de la media de amplitudes al cuadrado de los últimos 40 ms de la señal de QRS; QRSD: duración del QRS filtrado.
Nasir et al31 propusieron un nuevo criterio, que corresponde también a un retraso de la conducción en el miocardio del VD: la duración de la activación terminal del QRS ≥ 55 ms, medida desde el mínimo de la onda S hasta el final del QRS en V1, V2 o V3, en ausencia de BRDH completo. En su cohorte, este nuevo criterio menor estaba presente en el 95% de los pacientes con MAVD (incluido el 20% en que era la única anomalía del ECG detectada) y sólo en un 7% de los pacientes con taquicardia del TSVD y un 2% de los controles normales.
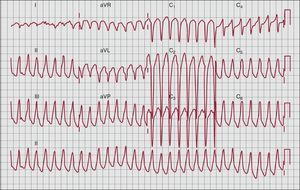
ArritmiasLas arritmias ventriculares son una observación frecuente en los pacientes con MAVD y a menudo constituyen la forma de presentación inicial de la enfermedad. La TV sostenida o no sostenida y las extrasístoles ventriculares tienen habitualmente una morfología de BRIH que refleja su origen en el VD (Figura 5). Cuando hay afección del VI, se puede observar arritmias ventriculares con una morfología de BRDH.

Figura 5. Taquicardia ventricular con bloqueo de rama izquierda del haz con eje inferior (origen del tracto de salida ventricular derecho).
El TSVD también es el lugar de origen de la TV del TSVD idiopática, una entidad benigna que se da en pacientes con un corazón estructuralmente normal. Los nuevos criterios de la MAVD lo tienen en cuenta para mejorar la especificidad de las arritmias ventriculares. En caso de TV con morfología de BRIH y un eje inferior (QRS positivo en las derivaciones II, III y aVF y QRS negativo en aVL), la TV constituye un criterio menor, puesto que puede observarse también en la TV de TSVD idiopática. En cambio, la TV con configuración de BRIH y un eje superior (QRS negativo o indeterminado en las derivaciones II, III y aVF y QRS positivo en aVL) es más específica de la MAVD y actualmente se considera un criterio mayor. Si no se conoce el eje de la TV, la arritmia se considera un criterio menor.
La presencia de más de 500 extrasístoles ventriculares en un registro de 24 h (que se ha reducido respecto a las 1.000/24 h de los criterios de 1994) constituye un criterio menor.
Antecedentes familiares y genéticaEl diagnóstico clínico de la MAVD en un individuo modifica el riesgo genético de sus familiares de primer grado, que pasa de 1:1.000-1:5.000 de la población general a 1:2. Los nuevos criterios de la MAVD lo reconocen al hacer que la presencia inequívoca de enfermedad en un familiar de primer grado constituya un criterio mayor (mientras que en los de 1994 era un criterio menor). Si el diagnóstico clínico de MAVD se da en un familiar de segundo grado o hay antecedentes de MAVD en un familiar de primer grado, pero no se puede determinar si dicho familiar cumple los criterios de la Task Force, se considera un criterio menor. Aplicando el mismo principio, los antecedentes de MAVD confirmada anatomopatológicamente en la autopsia o la cirugía constituyen un criterio mayor si se trata de un familiar de primer grado; en caso contrario, es criterio menor. Al igual que en los criterios de 1994, la muerte súbita cardiaca prematura de un individuo joven (< 35 años) a causa de una MAVD sospechada es un criterio menor.
Son cinco los genes que codifican proteínas desmosómicas a los que se atribuye la enfermedad en hasta un 70% de los pacientes32. Diversas mutaciones de genes no desmosómicos se han asociado a la MAVD, entre ellas, el gen del receptor de rianodina 2 (RYR2)11, mutaciones en las regiones no traducidas 5’ y 3’ del gen del factor de crecimiento transformador beta 3 (TGFβ3)12, la proteína transmembrana 43 (TMEM43)9, la desmina10 y los identificados más recientemente, el gen de la titina (TTN)13 y el de lamina A/C14. Los nuevos criterios de la MAVD incluyen la presencia de una mutación patógena asociada o probablemente asociada a la MAVD en un individuo con sospecha clínica de la enfermedad. La definición de la patogenicidad de la mutación es estricta y se basa en la demostración de que la variante no está presente o es muy poco común en las poblaciones de control sanas, altera o se predice que alterará la función y/o la estructura de la proteína codificada y/o se ha demostrado que se segrega junto con la enfermedad. Es esencial el seguimiento estricto de estos criterios, puesto que las variantes en genes desmosómicos parecen ser relativamente frecuentes en los individuos normales y su expresión es muy variable. De hecho, muchos pacientes presentan más de una variante patogénica, lo que indica que la carga genética es de especial importancia en esta enfermedad.
ConclusionesDe todas las miocardiopatías, la MAVD es la más difícil de describir de forma sencilla y clínicamente útil. Los nuevos criterios de la MVDA constituyen un avance en el intento de identificar a los individuos afectados por la enfermedad; aun así, la mayor parte de los diagnósticos reflejarán un grado variable de probabilidad, que dependerá en gran manera del contexto clínico. Más importe aún, hay que considerar los criterios modificados como un proceso dinámico, puesto que el conocimiento de la MAVD, su fundamento genético y los mecanismos patogénicos está en constante evolución.
Conflicto de interesesNinguno.
Autor para correspondencia: The Heart Hospital, 16-18 Westmoreland Street, Londres W1G 8PH, Reino Unido. perry.elliott@ucl.ac.uk