



El mayor reto que afronta el ser humano es la preservación de la salud. La única vía para generar mejores soluciones a los problemas de salud es la innovación, la verdadera innovación. La única fuente de auténtica innovación es la investigación, la investigación de calidad. El trayecto desde un estudio de investigación básica a un ensayo clínico aleatorizado es largo y no está libre de «baches» e incluso «minas». Estos son los obstáculos y las barreras que limitan la disponibilidad de recursos, dificultan el proceso administrativo-regulatorio y constriñen las iniciativas de los investigadores. Asistimos a una creciente demanda de evidencia que guíe la práctica clínica, pero paradójicamente acometer investigación biomédica se hace cada vez más complejo, caro y difícil de integrar a la práctica clínica, por el aumento de las barreras a la realización de los aspectos prácticos de la investigación. Nos enfrentamos al reto de aumentar el volumen de la investigación biomédica y al mismo tiempo mejorar su eficiencia y sus resultados. Este artículo revisa las diferentes etapas y modalidades de la investigación biomédica, desde los estudios no clínicos en modelos animales o computacionales a los ensayos aleatorizados y registros clínicos, centrándose en las limitaciones y los retos a los que se enfrentan, pero también aportando soluciones y alternativas que pueden ayudar a superarlos. Afortunadamente, los retos son siempre oportunidades disfrazadas.
Palabras clave
La investigación animal ha estado desde el principio en el centro de la controversia. En 1543, Andrés Vesalio publicó De humani corporis fabrica («Sobre la estructura del cuerpo humano»), y con ello no solo fundó la anatomía humana moderna como disciplina científica, sino que al mismo tiempo puso en duda la cuestión del valor de la anatomía comparada. Vesalio insistió en que el estudio de la anatomía humana requería la disección de seres humanos y no de especies próximas como los primates. Posteriormente hubo abundantes hallazgos anatómicos importantes, pero fueron pocos los avances que la investigación animal aportó a la ciencia médica. Alrededor de 230 años más tarde, el naturalista Stephen Hales describió la primera determinación de la presión arterial. En el volumen II de sus Statical Essays1, Hales explicó cómo introdujo tubos de latón en la arteria crural de una yegua inmovilizada, y cómo conectó un tubo de vidrio a los tubos de latón para que entrara la columna de sangre que ascendía, y así observar y registrar las oscilaciones de la columna como medida cuantitativa de la presión arterial. Sin embargo, no prosiguió en esta investigación y centró sus tendencias naturalistas en objetos menos animados como las plantas, ya que su vivisección fue muy criticada. Se dice que, en 1718, su buen amigo el poeta Alexander Pope, un renombrado amante de los perros, afirmó respecto a Hales: «Comete la mayoría de estas barbaridades con la idea de que sean de utilidad para el hombre. Pero, ¿cómo sabemos que tenemos derecho a matar criaturas de las que estamos tan poco por encima, como los perros, por curiosidad o aunque pueda sernos de alguna utilidad?»2.
Fue el siglo xix el que impulsó los trabajos realizados en animales, primero a través de los estudios pioneros del fisiólogo Claude Bernard y luego a través de las enigmáticas teorías de la evolución de Charles Darwin. En contra de las ideas entonces dominantes, Bernard insistió en que todas las criaturas vivas se rigen por las mismas leyes y de modo similar que la materia inanimada, y Darwin planteó la hipótesis de que el hombre descendía de otras formas de vida previas. Ambas líneas de pensamiento indican que se puede aprender mucho sobre procesos humanos a partir de la fisiología animal, ya que las fuerzas que los impulsan y las leyes de la naturaleza se mantienen incluso cuando la anatomía difiere. Las ideas de Bernard y de Darwin plantean que las verdades fundamentales de la condición humana podrían examinarse con más claridad y quizá hasta más pulcritud en sistemas animales vivos. Bernard realizó activamente estudios en animales, incluso en una época previa a la anestesia, y descubrió con su investigación las propiedades digestivas del páncreas, la función glucogénica del hígado, y el sistema vasomotor, con lo que creó el concepto de milieu intérieur (medio interno), al que más tarde Walter Cannon llamaría homeostasis. Por su ciencia y sus métodos, Bernard ha seguido siendo venerado hasta nuestros días por su uso de la vivisección, a pesar de que su esposa y su hija lo vilipendiaran. Darwin fue muy consciente y percibió el conflicto que conlleva comprender que sus teorías permitían, y hasta cierto punto estimulaban, el uso de la investigación en animales.
La aparición de la anestesia eliminó el aspecto evidente y fácilmente apreciable de la crueldad que suponía la ausencia de control del dolor y permitió alcanzar un mejor control sobre el estado de los animales y la reproducibilidad de los efectos. Hoy en día, el uso de un entorno controlado en la experimentación animal es esencial para el avance en los tratamientos médicos, incluida la optimización de medicamentos que salvan vidas, como la insulina, y la práctica totalidad de los dispositivos médicos que consiguen efectos relevantes. Sin embargo, esto no es porque los animales puedan ser un modelo de la enfermedad humana, puesto que no existen modelos de la enfermedad humana más que en el ser humano. No hay modelos animales de la enfermedad humana. Lo que sí es cierto es que los animales aportan un beneficio porque permiten hacer algo que rara vez puede hacerse en los ensayos clínicos en el ser humano: poner a prueba hipótesis sobre mecanismos de acción dentro de un marco de referencia preciso. En el estudio de la eficacia y la seguridad, aunque pueden obtenerse indicios en los animales, la demostración definitiva solamente puede hacerse en el ser humano. En cambio, se da la situación inversa por lo que respecta al estudio de los mecanismos. Las hipótesis acerca del modo de acción rara vez pueden confirmarse en las condiciones en que se encuentra el ser humano, que son extraordinariamente diversas; para ello son necesarios entornos controlados, en los que se pueda mantener contantes muchas condiciones y poner a prueba las ideas, es decir, son necesarios los estudios en animales. La experimentación animal es crucial porque es en estos seres vivos donde pueden validarse los conceptos fisiológicos que de otro modo nunca se podrían demostrar. Sin embargo, para que este estudio tenga valor, es necesario que se lleve a cabo únicamente si existe un compromiso absoluto con el respeto a los seres vivos y con la precisión en la realización de los experimentos. La utilidad de los estudios queda invalidada si se usan modelos inapropiados; por ejemplo, los que no permiten realizar comparaciones, los que emplean una fisiología fundamentalmente diferente, los que tienen un control inapropiado de los experimentos en animales, los que prestan una atención insuficiente o inadecuada a las necesidades de los animales y que no solo son crueles y éticamente inaceptables, sino que corrompen inevitablemente los resultados a causa de los estados de estrés no controlados, y los que no tienen luego un seguimiento en el ser humano. De hecho, una vez realizado un ensayo en animales y confirmados los resultados, deben llevarse a cabo ensayos en el ser humano para definir la seguridad y el efecto en comparación con lo propuesto. Esta es una advertencia importante y en modo alguno banal. De la misma manera que debe motivar una grave preocupación la realización de ensayos clínicos sin el apoyo de la información sobre el mecanismo de acción procedente de estudios en animales, debe considerarse también que un estudio realizado en animales es un desperdicio de recursos y una falta de respeto si no va seguido de ensayos clínicos. Así pues, no debe realizarse investigación en animales cuando no haya esperanza alguna de que vaya a tener repercusión traslacional de por sí o se prevea realizar en última instancia una validación clínica.
A este respecto, se plantean los dilemas modernos, como qué debe hacerse cuando los estudios en animales están tan lejos de la experiencia humana que requieren la creación de dispositivos o productos específicos para el animal, o cómo se debe valorar la actual prisa por realizar ensayos clínicos antes de haber comprendido completamente el efecto. En gran medida, se trata de las dos caras de la misma moneda. Por ejemplo, el estudio de válvulas cardiacas percutáneas se ve muy limitado por la notable diferencia anatómica entre el arco aórtico de cuadrúpedos como las ovejas o los cerdos y el del ser humano bípedo. Las válvulas que se pretende utilizar en el ser humano son extraordinariamente difíciles de introducir a través de la aorta de los primeros, en forma de campanario, lo que limita la utilidad del modelo animal. La insistencia en realizar estudios en animales con estos dispositivos podría requerir la creación de dispositivos que pudieran usarse únicamente en los animales. Una solución de este tipo es innecesaria y desvía la atención respecto al objetivo real. El desvío de la atención está en el desarrollo de un modelo que no es pertinente, pero el aspecto innecesario se debe a que el animal no es el único modelo no clínico y su inaccesibilidad no significa que no puedan validarse cuestiones operativas y mecánicas antes del uso en el ser humano. Se dispone de multitud de modelos que son aún más apropiados para realizar la valoración, así como de estudios in silico que pueden aportar una perspectiva conceptual antes de realizar ensayos en el ser humano. Rehuir estas alternativas preclínicas y decantarse en su lugar por la prueba prematura en el ámbito clínico constituyen una violación del método científico.
Al mismo tiempo, la prisa por realizar ensayos clínicos ha creado una serie de problemas que van en sentido contrario y pueden ser igualmente nocivos. La experiencia de los implantes endovasculares ha estimulado extraordinariamente la intervención cardiovascular, lo cual ha impulsado la biología vascular y la ciencia médica en paralelo con la innovación tecnológica. No obstante, la característica distintiva de esta experiencia es la atención profunda prestada a una evaluación preclínica precisa, multimodal y multidimensional, antes de realizar los ensayos fundamentales con fines de registro en el ser humano. La evaluación preclínica crucial de los stents metálicos precedió a los ensayos clínicos y a la evaluación y aprobación por la Food and Drug Administration (FDA) de Estados Unidos en 5 años, y la primera publicación definitiva de los resultados prometedores, y el modo de acción de los stents farmacoactivos apareció 5 años antes de la aprobación de estos dispositivos por la FDA3,4. Cuando surgieron problemas con estos dispositivos, hubo abundantes trabajos de experimentación animal, estudios de laboratorio y trabajos computacionales que permitieron orientar mejor la evaluación clínica. En cambio, no puede decirse lo mismo de la denervación renal ni, en menor medida, de los armazones bioabsorbibles. El ensayo Simplicity III HTN se realizó rigurosa y cuidadosamente y demostró de manera inequívoca la ausencia de efecto de la denervación renal por radiofrecuencia en comparación con los controles tratados con la intervención simulada5. Dada la fuerza de este ensayo, la mayoría de los programas de desarrollo de la denervación renal se han detenido y, sin embargo, la inclusión inicial en los ensayos, que se apoyó en los resultados prometedores de los primeros ensayos clínicos, se aceleró antes de que hubieran podido publicarse estudios definitivos en animales6,7. Solo en un examen retrospectivo se puede ver ahora cómo habría cambiado el enfoque clínico si se hubieran conocido de antemano los datos de los ensayos realizados en animales que aparecieron después de las publicaciones clínicas. Tal vez si estos artículos se hubieran publicado en primer lugar, no solo podríamos haber diseñado el ensayo de manera diferente, sino que también habríamos podido tener una respuesta menos binaria, de usar y luego dejar de usar, ante el hecho de que no se demostrara un efecto beneficioso. Esto es aplicable también al caso de los armazones bioabsorbibles. Las publicaciones iniciales en el campo de la evaluación preclínica de los implantes indicaban que los sistemas absorbibles requerían, en los animales, un periodo de observación mucho más largo que los dispositivos similares hechos de materiales duraderos, con objeto de tener en cuenta el tiempo previsto de erosión y eliminación de los materiales antes de que se iniciaran los ensayos clínicos8,9. Pero no se hizo así, y poco después de que datos clínicos repetidos demostraran que un armazón absorbible conlleva un aumento de las tasas de trombosis, la FDA emitió un comunicado de advertencia respecto al uso de este dispositivo. Solo ahora, tras la demostración clínica definitiva de un problema de seguridad, la trombosis, se aprecia que los resultados se atienen a lo que podría haberse predicho a partir de los modelos animales y computacionales de la erosión del material. El armazón absorbible, con el doble de superficie de material y el doble de grosor de los struts, muestra una tasa de trombosis temprana el doble de alta. Además, las tasas de trombosis continúan divergiendo entre el material erosionable y el duradero y alcanzan un nuevo máximo de manera tardía, coincidiendo con las condiciones inflamatorias que son necesarias para la absorción del material y en paralelo con esa absorción. Esto ilustra nuevamente un resultado clínico definitivo y una reacción de aceptar y luego dejar de aceptar, por lo que respecta a la fe en la tecnología, antes de haber obtenido un conocimiento pleno de la ciencia y la ingeniería en la que se basa el dispositivo. Nuestro temor es que los artículos de ciencia y de ingeniería no lleguen a publicarse o, si se presentan, no se lean con la atención y el cuidado que habrían generado si se hubieran publicado primero.
El problema que se produce cuando los ensayos clínicos no esperan a que se hayan realizado los trabajos en animales (es decir, la comercialización prematura de una tecnología) no solo expone a los participantes en los ensayos a una tecnología que se ha estudiado de manera incompleta en los animales, sino que, como ha ocurrido con la denervación renal y probablemente con los armazones bioabsorbibles, se elimina prematuramente también un conjunto de intervenciones que pueden ser prometedoras, antes de que se haya determinado de modo definitivo que tiene fallos. Solo el más intrépido o el menos informado seguirá respaldando una tecnología afectada por los fallos, aun cuando estos tengan su origen en una introducción errónea de ensayos clínicos en el proceso de desarrollo del producto. Se debe diferenciar aquí no solo un ensayo bien realizado de otro mal hecho, sino también el ensayo que se debe realizar del que no.
¿Dónde está el equilibrio? Ante todo, hay que reconocer que el diseño y el desarrollo terapéuticos son una cuestión de equilibrio: es preciso intentar que el conocimiento aumente en la mayor medida posible y pasar al ámbito clínico cuando ya no pueda ampliarse más la atenuación del riesgo. Es preciso reconocer también que no pueden hacerse afirmaciones absolutas a este respecto. No existe la seguridad absoluta, ningún dispositivo o fármaco puede ser enteramente eficaz y totalmente seguro; la idea de una eficacia completa se ve limitada por la noción de una seguridad absoluta. Hay una línea fina entre el efecto terapéutico más potente y la aparición de efectos secundarios indeseables. Al mismo tiempo, tenemos poco que ganar con un enfoque de «probemos, a ver qué ocurre». La mayoría de las veces, un abordaje de ese tipo está destinado al fracaso, y en caso tanto de éxito como de fracaso, no se sabe por qué se ha observado el resultado, por lo que hay pocas posibilidades de realizar una rectificación racional. En consecuencia, un enfoque integrado debe procurar primero formular y luego verificar una hipótesis sobre la acción, en el mayor número posible de ámbitos, como los experimentos de laboratorio multimodales y la modelización in silico, y luego el uso en modelos animales en un entorno muy controlado, con grados de complejidad creciente.
Un comentario final importante es que lo mejor es realizar ensayos clínicos con el mayor respaldo conceptual y de conocimiento del mecanismo de acción que sea posible, pero no es necesario haber completado todo el trabajo no clínico posible. No es el paso a los ensayos clínicos el enfoque clave principal del trabajo no clínico. Se trata más bien de que el trabajo no clínico motiva el paso a los ensayos clínicos como siguiente paso lógico para aportar una transformación en el tratamiento, el diagnóstico o la reclasificación de la enfermedad. De hecho, nosotros preferimos el término no clínico a preclínico, con objeto de no fomentar la idea de que la fase terminal de la investigación es la clínica. Hay una necesidad acuciante de que se realice investigación no clínica en paralelo con los ensayos clínicos y después de ellos. Las observaciones clínicas, como la de la trombosis con los armazones bioabsorbibles y la de la ineficacia de la denervación renal, hacen necesaria una mayor investigación no clínica. Cuando las observaciones clínicas no concuerdan con las expectativas existentes es precisamente cuando está justificado el estudio en el ámbito no clínico.
En resumen, el desarrollo de un dispositivo no puede ser completo sin el uso de modelos animales e incluso computacionales, puesto que solo en este ámbito no clínico se puede poner a prueba las hipótesis y explicar las observaciones. El entorno clínico permite demostrar el beneficio clínico y define la ventana de seguridad, pero son las investigaciones no clínicas las que son esenciales para explicar cómo y por qué funcionan los sistemas, así como los efectos de los cambios del entorno, las tensiones y las cargas sufridas. Seguimos dependiendo y estamos en deuda con estos sistemas y es preciso respetarlos y considerarlos con los mismos patrones de referencia científicos, éticos y técnicos que se emplean en los ensayos clínicos.
ENSAYOS CLÍNICOS ALEATORIZADOSLa creación de la medicina basada en la evidenciaDesde que se llevara a cabo el primer ensayo clínico aleatorizado (ECA) en 1948 (Medical Research Council Streptomycin Trial), los ECA han remodelado el conocimiento y la práctica médica, reduciendo el sesgo y mejorando la exactitud de la experimentación clínica. Los ECA han llegado a ser el patrón de referencia en la medicina basada en la evidencia. Estos ensayos han revolucionado la investigación médica y han mejorado la calidad de la asistencia sanitaria al esclarecer los beneficios y los inconvenientes de innumerables intervenciones10. Son los estudios que aportan la evidencia de mayor nivel para respaldar las recomendaciones de las guías y las autoridades reguladoras farmacéuticas los exigen para la autorización de medicamentos y dispositivos médicos, aunque con diversos grados de exigencia respecto a su calidad.
La importancia de la evidencia derivada de ECA se ha puesto de manifiesto claramente en una reciente investigación destinada a evaluar la coincidencia de los efectos terapéuticos en cuanto a la mortalidad observados en los registros y los posteriores ensayos aleatorizados. La evaluación incluyó 16 estudios observacionales elegibles y 36 ECA publicados posteriormente en los que se investigaron las mismas cuestiones clínicas11. En 5 (31%) de las cuestiones clínicas, el sentido de los efectos del tratamiento difirió entre los estudios observacionales y los ensayos clínicos. En 9 (56%) de los estudios observacionales, los intervalos de confianza no incluyeron la estimación del efecto obtenida en el ECA. En términos generales, los estudios observacionales mostraron unas estimaciones de la mortalidad significativamente más favorables (en un 31%) que los ensayos posteriores. Así pues, los registros podrían dar a las mismas cuestiones respuestas diferentes a las de un ECA posterior y es posible que sobrestimen considerablemente los efectos del tratamiento.
Parece claro que los ensayos aleatorizados amplios podrían seguir siendo necesarios para abordar cuestiones clínicas de importancia crítica para los resultados de interés para el paciente. Sin embargo, aunque los ECA en medicina cardiovascular han transformado la asistencia, en una revisión de 16 guías diagnósticas y de intervención específicas de enfermedades, solo un 12% de las recomendaciones fueron de nivel A (las más frecuentes para insuficiencia cardiaca, con un 26%, y las menos frecuentes para las valvulopatías, con un 0,3%), y un 48% de las recomendaciones fueron de nivel C12. Naturalmente, si no existen ensayos aleatorizados, los clínicos pueden actuar basándose en los resultados de los estudios observacionales, pero deben tener en cuenta que los efectos del tratamiento podrían ser menos claros. No obstante, las últimas 7 décadas han asistido también a muchas limitaciones de este «patrón de referencia» (tabla 1 y figura 1).
Ensayos clínicos controlados y aleatorizados
| Puntos fuertes |
| Si tienen un diseño correcto y potencia estadística suficiente, son el patrón de referencia |
| Eliminan el sesgo y el efecto de factores de confusión |
| Establecen la causalidad entre la intervención y el resultado |
| Retos |
| Requisitos regulatorios complejos |
| Organizaciones de investigación con ánimo de lucro contratadas |
| Múltiples autorizaciones éticas |
| Muchos parámetros no conocidos para el cálculo de la potencia estadística |
| No siempre tienen el tamaño suficiente, potencia estadística insuficiente para eventos «duros» (p. ej., muerte), potencia suficiente para objetivos combinados «duros ± blandos» (es decir, muerte ± reingreso) |
| El examen de selección previo es ineficaz e impredecible |
| Inclusión lenta (posible interrupción temprana) |
| Poblaciones muy seleccionadas debido a los criterios de exclusión (exclusión de poblaciones con alto riesgo y exclusión del uso fuera de las indicaciones autorizadas) |
| Aplicabilidad limitada |
| A menudo se realizan en centros de estudio especializados y seleccionados |
| A menudo utilizan objetivos sustitutos indirectos |
| Poco tiempo de seguimiento |
| Largo tiempo hasta la finalización y la realización completa |
| Coste muy elevado |
| A menudo solo los patrocina la industria (interés en nuevos tratamientos patentados y caros) |
| Con frecuencia se realizan después de la comercialización (disponibilidad de los resultados mucho tiempo después de la autorización) |
| La presentación puede ser deficiente (publicación tardía o nunca efectuada) |

Aun cuando los ECA han pasado a ser el método de referencia en la investigación farmacéutica, en las últimas décadas los investigadores clínicos se han esforzado en aplicarlos a otros campos de la medicina como, por ejemplo, la cirugía. Con la aparición de un mayor número de ECA en las décadas de los sesenta y los setenta, los cirujanos identificaron de manera creciente sus limitaciones: cada paciente tenía unas características patológicas únicas, cada cirujano tenía una pericia diferente y cada operación implicaba innumerables decisiones sobre la anestesia, la premedicación, el abordaje quirúrgico, la instrumentación y la asistencia posoperatoria, todo lo cual desafiaba la posibilidad de alcanzar la estandarización que requerían los ensayos clínicos. En las operaciones de cirugía mayor no podían usarse controles simulados, y ello limitaba la posibilidad de realizar ensayos clínicos con un diseño ciego13.
A veces, incluso los ECA bien realizados no han llegado a influir en la práctica médica. Las razones son múltiples, desde la presión del mercado hasta la inercia y el escepticismo. En 2002, el ensayo ALLHAT reveló que los diuréticos tiacídicos genéricos eran igual de eficaces que los antagonistas del calcio y los inhibidores de la enzima de conversión de la angiotensina, de aparición más reciente y de coste elevado, en el tratamiento de la hipertensión14. Sin embargo, las ventas de estos antihipertensivos más recientes aumentaron con mayor rapidez que las de los diuréticos.
El uso del balón de contrapulsación intraaórtico no ha disminuido como se esperaba tras la aparición del ensayo de referencia que mostró que no aportaba beneficio alguno en cuanto a la mortalidad15.
Por otro lado, los resultados de algunos ECA se han aceptado como un hecho, pero luego se ha demostrado que carecían de validez externa. Las inquietudes sociales y éticas han puesto en duda también la legitimidad de algunos ECA. Algunos autores defienden el empleo de enfoques más flexibles para la investigación clínica, como el uso de objetivos sustitutos indirectos, la autorización condicionada de las autoridades reguladoras y las vías paralelas para permitir el acceso a los medicamentos fuera de los ensayos clínicos. Sin embargo, a los críticos con este planteamiento les preocupa que unas normas menos estrictas puedan socavar el rigor científico y fomentar un proceso arriesgado de desregulación dominado por la industria farmacéutica.
Una inquietud que se viene produciendo desde hace mucho tiempo es la discrepancia entre el marco temporal en que tienen lugar los ECA y el rápido avance de la innovación. Justo cuando se ha acumulado datos suficientes durante un periodo de tiempo suficiente, resulta que determinada técnica quirúrgica/intervencionista ha mejorado, que un tratamiento médico ha cambiado o con ambas cosas, y las conclusiones han dejado de ser aplicables.
Sin embargo, los ECA están a «precio de oro»Los ECA han pasado a ser cada vez más amplios, complejos y costosos, lo que puede poner en peligro su propia existencia16. Actualmente un solo ECA de fase 3 podría llegar a costar 30 millones de dólares o más. Un ensayo amplio con 14.000 pacientes inscritos en 300 centros tiene un coste de 300 millones de dólares17. Sin embargo, el coste no es el mismo en todo el mundo, y Estados Unidos es el país con mayor coste, mientras que este es un 50% inferior en Alemania, un 39% inferior en Polonia y un 36% inferior en India. Por consiguiente, los diversos países compiten actualmente por convencer a la industria farmacéutica y las organizaciones de investigación bajo contrato de que sus perfiles de regulación farmacéutica, asistencia clínica y salud pública proporcionan las condiciones ideales para la realización de los ensayos, a pesar de que sea improbable que los productos evaluados lleguen a estar disponibles para las poblaciones locales después de finalizado el ensayo18.
Con el desarrollo de los ECA hasta convertirse en instrumentos de comercialización de alto coste y alto valor, floreció una industria de los ensayos clínicos. Tras su surgimiento a finales de la década de los setenta, las organizaciones de investigación bajo contrato han pasado a ser una industria con grandes beneficios19.
Errores en el diseño y la presentación de los ECANo es infrecuente que el diseño y la presentación de los ECA sean deficientes. De los 96.346 estudios registrados en ClinicalTrials.gov entre 2007 y 2010, la mayoría son de pequeño tamaño y tienen métodos de presentación heterogéneos20. De los 13.327 ensayos registrados entre 2008 y 2013, solo un 13% presentaron resultados en un plazo de 12 meses tras su finalización21. De entre 244 ensayos externos financiados por el National Heart, Lung, and Blood Institute que se completaron entre 2000 y 2011, solo el 64% se había publicado al llegar 2012 y la mediana de tiempo hasta la publicación fue de 25 meses22.
Los resultados del ensayo REVIVE, en el que se evaluó el sensibilizador del calcio levosimendán en pacientes con insuficiencia cardiaca aguda descompensada, se publicaron más de 7 años después de finalizado el estudio y, sin embargo, el fármaco se había comercializado ya en más de 40 países23.
Se han elaborado guías para mejorar la calidad da la presentación de los ECA. Se ha demostrado empíricamente que la aplicación y el respaldo a algunas de estas guías, como la declaración CONSORT, han mejorado la calidad de la presentación de los estudios24–26.
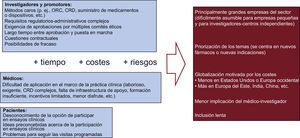
Obstáculos y oportunidades para los ECADe los retos y obstáculos existentes para la realización de los ECA (figura 1), el coste continúa siendo el más importante. Los costes extraordinariamente altos de los ensayos grandes solo están al alcance de empresas grandes interesadas en sus nuevos fármacos o dispositivos. Las prioridades en las preguntas de investigación que establecen las empresas son diferentes de las que tiene la sociedad en general, que prioriza la comparación de los tratamientos de uso frecuente.
Los complejos procedimientos administrativos y de regulación requieren tiempos muy largos y los riesgos de que el estudio fracase deben ser asumidos por su promotor. Estos múltiples obstáculos pequeños son especialmente engorrosos en el caso concreto de los ensayos iniciados por investigadores.
Por otro lado, los médicos afrontan múltiples problemas por lo que respecta a su participación activa (principalmente de los centros a los que pertenecen). Por último e igualmente importante, los pacientes no son conscientes de la opción de participar o muchos de quienes lo son optan por no hacerlo a causa de ideas preconcebidas sobre la seguridad y la privacidad.
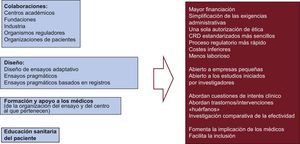
Por fortuna, existen alternativas para superar la mayoría, si no la totalidad, de estos obstáculos (figura 2). La más importante es fomentar la colaboración activa nacional y supranacional entre instituciones académicas y sanitarias, industria, fundaciones dedicadas a enfermedades concretas, grupos de pacientes y organismos administrativos-gubernamentales. En los últimos años, algunos ensayos grandes en el campo de la medicina cardiovascular, como el DAPT, se han financiado mediante la colaboración entre instituciones académicas e industria27. Estas colaboraciones se han puesto en marcha para abordar los retos terapéuticos de las denominadas enfermedades huérfanas, como la fibrosis quística o la diabetes melitus tipo 128. Puede observarse una priorización coordinada de las necesidades de investigación en el interés creciente de las administraciones sanitarias de algunos países por la investigación comparativa de la efectividad.

Otras alternativas importantes son las que se basan en los diseños de ensayos adaptativos, el apoyo a los médicos en los centros a los que pertenecen y los programas de educación sanitaria de los pacientes. Por lo que respecta al enfoque de un diseño adaptativo, este método puede aplicarse a los ensayos clínicos exploratorios (determinación de las dosis seguras y eficaces o modelos de la relación dosis-respuesta) o a los de confirmación (con la introducción planificada de manera prospectiva de la evolución futura de un ensayo en curso, basándose en el análisis de los datos acumulados en el propio ensayo). Hay ejemplos de ensayos en los que se han hecho nuevas estimaciones del tamaño muestral o se ha modificado el objetivo principal29,30.
Otra alternativa para el diseño adaptativo es la derivada del uso de diseños de «enriquecimiento» de la población empleando biomarcadores. Existe la posibilidad de que con biomarcadores predictivos se identifique a los pacientes que es probable que obtengan un efecto beneficioso con los tratamientos dirigidos y, por consiguiente, se aumente la tasa de éxitos en los ensayos clínicos de confirmación. Empleando este diseño, se incluye en la asignación aleatoria a todos los participantes, con independencia de sus valores de biomarcadores, pero se emplea un análisis provisional para determinar si el beneficio obtenido en los pacientes positivos para un biomarcador es distinto del observado al ensayar el producto en pacientes negativos para el biomarcador. Si el efecto beneficioso se da solo en los pacientes positivos para el biomarcador, se detiene la inclusión de nuevos pacientes en el subgrupo de los negativos para el biomarcador. El análisis estadístico final de los datos se basa entonces en los datos de las 2 etapas, empleando métodos de prueba cerrados y de tasa de error condicional, con objeto de no incrementar el error de tipo I31.
MetanálisisEn los últimos años se ha asistido a un aumento exponencial del número de metanálisis publicados. No hay ningún tema de la terapéutica cardiovascular que carezca de su correspondiente metanálisis. De hecho, a lo largo de un año cualquiera, se publican varios metanálisis que abordan el mismo tema.
El beneficio que aporta este enfoque es la combinación de los estudios (ensayos clínicos y estudios observacionales), que conduce a una mayor potencia estadística y a estimaciones puntuales más sólidas que las posibles con los datos aportados individualmente por cada estudio. Sin embargo, al llevar a cabo un metanálisis, el investigador debe tomar decisiones que afectan a los resultados, como los criterios de búsqueda, la forma de tratar los datos incompletos o los métodos estadísticos que deben usarse.
Es importante conocer las limitaciones que tiene este método. Un metanálisis de varios estudios pequeños no predice los resultados de un solo estudio más amplio. Un buen metanálisis no permite corregir el diseño incorrecto o el sesgo que puedan tener los estudios de origen. En un metanálisis deben incluirse únicamente los estudios metodológicamente sólidos (síntesis de la mejor evidencia)32.
Otro posible error es el derivado del sesgo de publicación o «gaveta de archivos». Esto ocurre como consecuencia de basarse en los estudios publicados, que pueden mostrar unos resultados exagerados a causa del sesgo de publicación, ya que los estudios que muestran resultados negativos o no significativos es menos probable que sean publicados33. Además, las declaraciones de intereses no siempre se notifican de manera completa y hay un posible problema derivado del sesgo causado por la planificación. Otros puntos débiles son los derivados de la metodología estadística, ya que falta por determinar el método estadísticamente más exacto para analizar los resultados combinados. A este respecto, están aumentado las críticas al popular modelo de efectos aleatorios.
En la declaración PRISMA se presentan directrices que establecen un conjunto mínimo de elementos basado en la evidencia para la presentación de las revisiones sistemáticas y los metanálisis34.
Ensayos aleatorizados pragmáticosDebe establecerse una distinción entre los ECA en función de su enfoque principal. Los ensayos explicativos o de mecanismo de acción se centran en la pregunta «¿Esta intervención puede dar resultado en condiciones ideales?», mientras que los ensayos pragmáticos intentan responder a «¿Esta intervención da resultado en las condiciones habituales?»35.
Se ha elaborado una herramienta que establece un conjunto de criterios para ayudar a los investigadores a determinar en qué medida su ensayo es pragmático o explicativo. La herramienta se denominó inicialmente PRECIS (Pragmatic-Explanatory Continuum Indicator Summary) y dispone actualmente de una nueva versión, PRECIS-236,37. Se ha propuesto, además, una ampliación para ensayos pragmáticos de la declaración CONSORT38.
Los ensayos pragmáticos están diseñados para mostrar la efectividad en las condiciones de «práctica clínica real» de las intervenciones en amplios grupos de pacientes e identificar los subgrupos a los que la innovación aportará el máximo beneficio. En consecuencia, estos ensayos se caracterizan por un gran tamaño muestral, poblaciones representativas y resultados relevantes, un uso eficiente de los recursos existentes, operaciones simplificadas (monitorización limitada, notificación de la seguridad, evaluaciones específicas del ensayo y documentación del cumplimiento y de las cuestiones sobre regulación farmacéutica), obtención de datos basales y, si es posible, de los resultados incluida en el contexto de la asistencia ordinaria o mediante seguimiento telefónico o automático, aprovechamiento de las historias clínicas electrónicas (HCE) y los registros, y unos formularios simplificados de consentimiento informado y de recogida de datos de los pacientes39–41.
Las dimensiones clave que definen un ensayo pragmático se centran en la inscripción de pacientes e investigadores, el tipo de intervenciones y el modo de aplicarlas, el tipo de seguimiento y el tipo y la determinación de los parámetros de valoración.
En los ensayos pragmáticos, los participantes deben ser similares a los pacientes a los que se aplicará la intervención si llegara a convertirse en la forma de asistencia habitual. Los criterios de inclusión y exclusión deben reducirse al mínimo, y es preciso optimizar el número y la complejidad de las visitas y los procedimientos aplicados en el estudio. En algunos contextos, es posible incluso obviar la necesidad del consentimiento informado. Esto es lo que sucede cuando se aplica la aleatorización por grupos, la cual incluye el empleo de grupos de pacientes (en el mismo centro sanitario) a los que se ha asignado aleatoriamente la misma intervención42.
Un enfoque relacionado es el diseño de aleatorización múltiple de cohortes, en el cual se inscribe una cohorte de participantes y se obtiene su consentimiento para el seguimiento y su posible inclusión en ensayos clínicos de nuevos tratamientos en comparación con la asistencia estándar43.
Por lo que respecta a los investigadores, en los ensayos pragmáticos deben participar diversos tipos de investigadores, con una combinación representativa de la experiencia apropiada para la aplicación de la intervención en estudio. Si es probable que haya cierta heterogeneidad en las respuestas a la intervención, el ensayo debe ser lo bastante grande para permitir una interpretación de esa heterogeneidad. La creación de redes clínicas, el establecimiento de comunidades de investigación para enfermedades específicas y el reconocimiento a los profesionales de la salud por su investigación son elementos de clara utilidad.
La aplicación de la intervención debe ser tan próxima a la práctica clínica normal como sea posible. Esto implica que los ensayos pragmáticos utilizan con frecuencia un diseño sin enmascaramiento. Por consiguiente, la notificación de los acontecimientos adversos no graves, las razones del abandono del tratamiento y diversos parámetros de valoración evaluados por los pacientes están sujetos a un mayor grado de sesgo, lo cual afecta a la calidad del ensayo. Para reducir al mínimo estos sesgos, es importante centrar el análisis de los parámetros de valoración en los eventos importantes (como muerte o ingreso hospitalario en urgencias).
Lo ideal es que el seguimiento se realice empleando las HCE. Esta estrategia solo es viable en los sistemas de asistencia sanitaria que disponen de una HCE fiable, estandarizada y accesible que recoge los eventos de interés. La identificación de los pacientes que están incluidos ya en registros específicos de una enfermedad o de una intervención brinda una oportunidad eficiente y de bajo coste de realizar ensayos pragmáticos, como el TASTE del registro cardiovascular de Suecia44. Se han llevado a cabo muchos ensayos con este enfoque pragmático, pero la mayoría de ellos no han utilizado registros, que tienen la ventaja adicional de brindar datos iniciales y, en algunos casos, de resultados, incluidos en la asistencia clínica habitual o accesibles a través de una vinculación automática de las fuentes de datos45.
Por lo que respecta a los parámetros de valoración, los diseños de ensayo pragmático podrían tener más limitaciones para evaluar objetivos que requieran la aplicación de métodos que no forman parte de la práctica clínica ordinaria. La presuntamente innecesaria validación («adjudicación») de los eventos en los ensayos pragmáticos es una cuestión discutible, puesto que dicha validación corresponde más a un tema de calidad que a una cuestión pragmática.
La diferencia entre los ensayos explicativos y los pragmáticos podría expresarse, de manera algo simplista, como una mayor validez interna de los estudios explicativos frente a una mayor validez externa de los ensayos pragmáticos. Es importante tener en cuenta que las características de los ensayos pragmáticos que respaldan la posible generalización o aplicabilidad de sus resultados, como son la inclusión de poblaciones de pacientes heterogéneas, la ausencia de enmascaramiento, la ausencia de un grupo placebo o una deficiente adherencia al tratamiento, pueden reducir también la sensibilidad y limitar la interpretación de los resultados. No a todas las cuestiones clínicas se puede responder con un diseño de ensayo pragmático; por lo tanto, este enfoque debe aplicarse siempre que sea viable y cuando no comprometa la calidad del ensayo y la posibilidad de dar respuesta a la pregunta clínica de interés41.
Ensayos aleatorizados basados en registros pragmáticosComo se ha mencionado antes, las HCE y los registros clínicos de calidad cardiovascular brindan oportunidades para la realización de ECA prospectivos pragmáticos y basados en los registros. El empleo de procedimientos simplificados de regulación farmacéutica, ética y consentimiento, la inclusión integrada en la asistencia de la «práctica clínica real» y los sistemas simplificados o automáticos de obtención de datos basales y de resultados permiten evaluar la potencia y la viabilidad del estudio, una inscripción rápida y eficiente, la presentación de resultados generalizables a un bajo coste y un posible uso novedoso y basado en la evidencia de fármacos genéricos, con un coste bajo para la sociedad46.
El registro SWEDEHEART de Suecia es la plataforma del primer ECA basado en un registro (TASTE) y, más recientemente, esto se ha aplicado también en el iFR-SWEDEHEART44,47. Asimismo el American College of Cardiology ha colaborado con investigadores afiliados (y ha contado con el apoyo del National Heart, Lung, and Blood Institute) para dirigir el uso del registro CathPCI como sistema de base de obtención de datos de un ECA en el que se ha comparado el acceso radial con el femoral en mujeres a las que se practica un cateterismo cardiaco48.
Las características peculiares del registro sueco se comentan en el siguiente apartado de esta revisión, pero es crucial facilitar la realización de este tipo de ECA. La posibilidad de extraer datos tanto basales como de resultados a partir de registros de calidad vinculados con bases de datos administrativas es clave para la realización de estos ensayos.
Los ECA pragmáticos basados en registros brindan sus propios beneficios y posibles limitaciones (tabla 2). Las ventajas más relevantes son las relativas a la capacidad de incluir muestras representativas mucho más amplias en un corto periodo y con unos costes significativamente inferiores. Además, dado que la monitorización se reduce al mínimo, las limitaciones más importantes son las relativas a la calidad de los datos, la uniformidad de la validación de los eventos y mantener la privacidad.
Ensayos clínicos aleatorizados pragmáticos (basados en registros)
| Puntos fuertes |
| Evidencia basada en estudios aleatorizados en la práctica clínica real |
| Proceso de regulación simplificado |
| Organizaciones de investigación académicas sin ánimo de lucro |
| Una sola aprobación ética |
| Tamaño de la muestra con una potencia estadística adecuada para eventos de interés, muestra apta para el estudio y tasas de eventos conocidas |
| El examen de selección previo es automático, eficiente y predecible |
| Poblaciones de pacientes no seleccionadas-generalizables: eficacia y efectividad |
| Gran número de eventos, lo que permite identificar los eventos muy poco frecuentes |
| Evaluación automática de los parámetros de valoración |
| Menor coste |
| Fomentan la aplicación de la evidencia a la práctica clínica |
| Evalúan los usos nuevos de fármacos/dispositivos ya existentes |
| Puntos débiles |
| Calidad de los datos |
| Validación de los eventos |
| Enmascaramiento respecto a las intervenciones |
| Se reduce al mínimo la monitorización, lo cual tiene implicaciones en cuanto a la seguridad de los participantes en el ensayo |
| Privacidad en estas bases de pacientes amplias |
| Equilibrio entre eficacia y efectividad |
| Evaluación de los efectos en parámetros de valoración percibidos por los pacientes, como la calidad de vida |
| Realización multinacional |
Pese a que los ECA basados en registros son mucho más baratos que los ECA tradicionales, siguen implicando un coste considerable, que generalmente supera las posibilidades de las subvenciones recibidas por los centros o los investigadores. Tiene especial importancia que las diversas partes interesadas, como los organismos públicos de regulación farmacéutica y de financiación, identifiquen la necesidad de reformar los ensayos y la conveniencia de financiar ensayos pragmáticos. Ha llegado el momento de que tanto la industria como los financiadores públicos aprovechen estas nuevas formas de realizar ECA basadas en su eficiencia y su bajo coste para llegar a nuevos tratamientos para los pacientes, junto con un ahorro para esas partes interesadas y la población49–53.
REGISTROS CLÍNICOSUn registro clínico es una base de datos observacional centrada en un trastorno clínico, un tratamiento o una población. Se recogen de manera sistemática los datos con unos fines científicos, clínicos o de decisión política especificados. En los registros clínicos, no se aplica un abordaje terapéutico obligatorio y los criterios de inclusión son amplios, junto con pocos criterios de exclusión. Los registros clínicos se centran en captar los datos que reflejan la «práctica clínica real» en poblaciones de pacientes amplias y representativas54.
Los registros pueden clasificarse en función de las características de la población incluida o según su finalidad (medición de la calidad, investigación o fines múltiples). Pueden ser de diseño prospectivo o retrospectivo y los llevan a cabo muchos tipos de entidades (investigadores, asociaciones profesionales, organizaciones sin ánimo de lucro, organismos de la administración e industria). Los registros clínicos recogen los datos utilizando definiciones y normas estandarizadas. Aunque generalmente los datos proceden de resúmenes de historias clínicas y de formularios de recogida de datos, algunos registros los extraen directamente de las HCE generalizadas.
Resultados y calidad de la asistencia sanitariaEn los últimos años ha habido cada vez más énfasis en la medición y la mejora de la calidad y la eficiencia de la asistencia médica y, en consecuencia, una proliferación de registros clínicos diseñados para evaluar la aplicación y los resultados de la asistencia en un contexto de «práctica clínica real». Los registros clínicos constituyen un elemento clave para medir los resultados de los procesos de asistencia y aportan una retroalimentación útil a los profesionales de la salud, así como una mejora de la calidad de la asistencia. La evaluación de la prestación de la asistencia sanitaria se hace cada vez más por medio de registros, entre los que se encuentran los siguientes55:
- •
Evaluación de la efectividad y la seguridad de la asistencia sanitaria.
- •
Medición de la idoneidad de la asistencia y de las disparidades en su prestación.
- •
Medición de la efectividad de la mejora de la calidad.
- •
Evaluación de los factores que influyen en el pronóstico y la calidad de vida.
- •
Mejora de los resultados clínicos, la experiencia en la asistencia de pacientes y los resultados percibidos por estos.
Un estudio recientemente publicado aporta datos del año 2014 de 4 programas de calidad hospitalaria del National Cardiovascular Data Registry de Estados Unidos, que muestran las tendencias en la asistencia cardiovascular56.
Todos los agentes involucrados en la asistencia sanitaria obtienen un beneficio con los registros:
- •
En el caso de los profesionales de la salud, los registros mejoran la calidad de su asistencia y los resultados clínicos de los pacientes y permiten aplicar una medicina personalizada.
- •
Por lo que respecta a los pagadores de la asistencia, los registros evalúan la calidad y el coste que supone (relación coste-efectividad).
- •
En el caso de la industria, los registros demuestran el valor de los productos y llevan los nuevos fármacos, dispositivos y servicios al mercado más rápidamente y con mayor probabilidad de que tengan valor clínico y económico.
- •
En lo relativo a los pacientes, los registros implican una participación más activa en el proceso de gestión de la salud y permiten una interacción dinámica con los prestadores de la asistencia sanitaria, lo cual facilita la adherencia al tratamiento y fomenta las conductas saludables.
Indudablemente, el patrón de referencia para la generación de la medicina basada en la evidencia es los ECA. La cardiología es verdaderamente una especialidad basada en la evidencia, aunque en realidad solo una minoría de las recomendaciones de las guías de práctica clínica están respaldadas por evidencia del nivel de calidad más alto12. En muchas cuestiones clínicas, no se han realizado ECA adecuados y es muy probable que nunca lleguen a realizarse, principalmente por consideraciones éticas y económicas. Las intervenciones no relacionadas con la introducción de nuevas patentes de fármacos, dispositivos y servicios (intervenciones huérfanas) son poco evaluadas en los ECA debido a la falta de interés por su financiación.
Como ya se ha comentado, aunque los ensayos aleatorizados se consideran el patrón de referencia para comparar la efectividad, tienen algunas limitaciones (tabla 1). La principal es que los ECA son cada vez más costosos y que en general la inclusión es restringida, lo cual conlleva problemas para la generalización de los resultados.
Inclusión restringida y aplicabilidad de los ECASe estima que menos de un tercio de los pacientes con insuficiencia cardiaca de la práctica clínica cumplirían las condiciones exigidas para la inclusión en los ECA y que a cerca de dos tercios de los participantes en la Euro Heart Survey on Coronary Revascularization no se los habría considerado aptos para participar en ECA de comparación entre intervención coronaria percutánea y cirugía de bypass arterial coronario57–59. Los pacientes de la práctica clínica eran de más edad y tenían mayor probabilidad de sufrir comorbilidades. Aunque los ECA no mostraron diferencias entre la intervención coronaria percutánea y la cirugía de bypass arterial coronario en los criterios de valoración del resultado, en los pacientes no aptos para la inclusión en ensayos, se registró un beneficio claro en la supervivencia a 1 año con la intervención coronaria percutánea en comparación con la cirugía de bypass arterial coronario59. Los resultados contrarios de un análisis de un registro de Nueva York podrían explicarse por este contraste entre los pacientes aptos y no aptos para los ensayos60.
Los registros prospectivos se emplean para determinar si los resultados de los ECA realizados en poblaciones seleccionadas pueden trasladarse a la población clínica general. Si los pacientes con mayor riesgo no están suficientemente representados en los ECA, los registros desempeñan un papel importante para la validación de los resultados de ensayos en grupos de población excluidos o infrarrepresentados. El análisis de estos subgrupos a través de registros es de capital importancia, dado el aumento de la tasa de complicaciones que se da en tales pacientes y la posibilidad de que los tratamientos causen un daño. A este respecto, se excluye sistemáticamente de los ECA a los pacientes de edad avanzada o se les da mínima representación61.
Potencia estadística escasa y poco alcance temporal de los ECALos eventos adversos que se producen de manera muy tardía o con escasa incidencia pueden pasar inadvertidos fácilmente en los ECA, debido al tamaño muestral insuficiente o el corto periodo de seguimiento. Esto es lo que ocurrió con los episodios muy tardíos de trombosis de los stents farmacoactivos y los ensayos clínicos llevados a cabo con los stents farmacoactivos de primera generación62. Los registros con poblaciones más amplias y periodos de seguimiento más largos permitieron identificar el aumento del riesgo asociado63,64.
Esto tiene especial importancia respecto a los nuevos medicamentos, con los cuales es posible que se produzcan efectos adversos de muy baja frecuencia. Tras la farmacovigilancia poscomercialización, es crucial establecer la seguridad de los nuevos tratamientos que se autorizan con base en ensayos con relativamente poca potencia estadística y seguimiento corto. Además, los registros pueden hacer que surjan motivos de preocupación respecto a la seguridad que no se hayan detectado en los ensayos previos, no como consecuencia de sus limitaciones, sino simplemente por que no se han realizado adecuadamente o no hayan finalizado aún, o a causa de que el tiempo transcurrido desde la autorización del dispositivo sea insuficiente. Este es el caso del riesgo de trombosis con los armazones basculares bioabsorbibles65.
Limitaciones de los registros para la generación de evidencia médicaLos puntos fuertes y débiles de los registros se presentan en la tabla 3. Los registros prospectivos no son aleatorizados, por lo que los resultados sobre la eficacia del tratamiento deben interpretarse con precaución. La presencia de pequeños desequilibrios cuando hay factores de confusión no medidos que tienen una intensa relación con los resultados evaluados del tratamiento puede tener repercusiones importantes al introducir una confusión en la relación entre tratamiento y resultados. Como ejemplo de la posible repercusión de factores de confusión no previstos, cabe citar que el análisis inicial de la cohorte SCAAR de 2003 a 2004 indicó un aumento de las tasas de mortalidad asociadas a los stents farmacoactivos en comparación con los stents metálicos que se invirtió en el análisis de la cohorte de 2003 a 200666,67. Esta inversión de los resultados se atribuyó a un mejor equilibrio de los 2 grupos en cuanto a las características de la lesión y del stent en este último periodo de estudio.
Registros clínicos
| Puntos fuertes |
| Poblaciones de pacientes no seleccionadas-generalizables |
| Poblaciones muy amplias |
| Caracterización epidemiológica |
| Gran número de eventos, lo que permite identificar eventos muy poco frecuentes |
| Eficientes |
| Bajo coste |
| Posibilidad de un seguimiento longitudinal |
| Posibilidad de aplicación de características de diseño dinámicas durante la realización |
| Resultados comparados, generación de hipótesis |
| Puntos débiles |
| No valoran la efectividad comparada, muy limitados para la investigación comparativa de resultados |
| Imposibilidad de introducir ajustes por factores de confusión, pese a usar modelos estadísticos avanzados |
| Ausencia de enmascaramiento respecto a las intervenciones |
| Asegurada una estricta inclusión no consecutiva (sesgo de inclusión) |
| Calidad de los datos variable y cuestionable |
| No hay un proceso de validación de los eventos uniforme ni centralizado |
| Los prejuicios o las ideas preconcebidas acerca de los efectos del tratamiento pueden sesgar la notificación de los eventos |
| Auditorías escasas o inexistentes |
El sesgo se puede atenuar, aunque nunca se llegue a eliminarlo por completo, mediante el diseño o el análisis. Por lo que respecta al diseño, se puede aplicar criterios de emparejamiento para la inclusión/exclusión y usar la disponibilidad emparejada (cronológica o geográfica). En la fase de análisis, el uso de ajustes por covariables, emparejamiento de casos y puntuaciones de propensión, junto con otros métodos estadísticos actualmente existentes, puede resultar de utilidad68,69.
Teniendo en cuenta las mencionadas limitaciones de los registros, una de las principales funciones claramente establecidas de estos es la generación de hipótesis, con objeto de proponer ECA que puedan confirmar o rechazar lo observado en los registros. Como ejemplo de esto, cabe mencionar que en los registros el tratamiento estrogénico en las mujeres mostró un efecto beneficioso en resultados clínicos cardiovasculares, pero no en un ensayo clínico adecuado70,71.
Las bases de datos amplias pueden utilizarse para señalar la inclusión en ECA de manera individualizada a través de métodos de análisis de factores que identifiquen aquellos que hacen que los pacientes sean comparables en cuanto al pronóstico.
Como ya se ha comentado en esta revisión, los registros clínicos pueden usarse como plataforma para el desarrollo de ensayos aleatorizados y la investigación comparativa de la efectividad, que podrían alcanzar el doble objetivo de reducir los costes totales del ensayo y simultáneamente aumentar las posibilidades de generalización de los resultados.
Claves de un registro satisfactorioLa medición adecuada de los resultados clínicos requiere una terminología clínica estandarizada, unas normas uniformes para la definición y la obtención de los datos, estrategias para adaptarse a la complejidad del paciente, técnicas para verificar si los datos son completos y exactos y obtener datos longitudinales72,73. Las características clave de un registro de alta calidad y los nuevos avances que se produzcan pueden delimitarse de manera escalonada (tabla 4)74.
Características clave de un registro satisfactorio
| Comité directivo e investigador principal designados desde el comienzo |
| Métodos de revisión ética adecuados |
| Definiciones estandarizadas para la obtención de datos y la notificación de eventos |
| Selección aleatorizada de los centros (idealmente, participación del 100%) |
| Inclusión de pacientes consecutivos para asegurar la representatividad |
| Captación electrónica de los datos, con explicaciones claras y sencillas de las definiciones y las instrucciones a los participantes y controles plausibles para detectar los datos introducidos de manera incorrecta |
| Herramientas integradas para un feedback rápido a los centros participantes |
| Compilación de los datos y análisis estadístico centralizados, a cargo de estadísticos profesionales |
| Auditoría de como mínimo un pequeño grupo de centros elegidos al azar |
| Presentación de todos los datos recogidos, con conclusiones apropiadas para estudiar el diseño |
| Notificación transparente de los investigadores y las fuentes de financiación en todas las publicaciones |
| Perspectivas futuras: |
| • Aumento de la interoperabilidad, que permita la gestión de la gobernanza/flujo de trabajo |
| • Aplicación de métodos sólidos de aprendizaje automático (machine learning) |
| • Internet de las cosas: menos dependencia de la entrada manual de los datos, nuevas fuentes de datos, tecnología de biosensores llevables y complementos inteligentes para el seguimiento automático y ecosistemas digitales |
| • Determinación del fenotipo multidimensional de los pacientes: sistema de información geográfica médica, de la demografía a la «-ómica» en general |
El aumento en el uso de las HCE es a la vez un reto y una oportunidad por lo que respecta al posible uso de los registros clínicos. Dado que los hospitales utilizan cada vez más las HCE, es preferible la extracción automática y directa de la HCE de algunos datos del registro, en vez de tener que introducirlos manualmente. En última instancia, esta estrategia ahorra tiempo y dinero. La interrelación entre la HCE y los registros clínicos es una situación en la que todos salen ganando. Las HCE y los datos administrativos podrían facilitar el seguimiento longitudinal de los pacientes y la captación de los resultados no clínicos, como el uso de recursos. A su vez, los registros podrían aportar la disciplina de los modelos de datos comunes y las definiciones de datos sistemáticas y la calidad de los datos a las HCE.
Sin embargo, hay algunas preocupaciones importantes al respecto55:
- 1.
La entrada de datos en registros clínicos con un objetivo específico la realizan profesionales que extraen la información de la historia clínica aplicando unas definiciones específicas. En cambio, los datos que constan en las HCE se obtienen durante el proceso de la asistencia al paciente por diversos profesionales de la salud, con fines distintos del análisis y la notificación.
- 2.
La extracción automática de algunos datos es viable, como los resultados analíticos y los parámetros demográficos, pero extraer de la HCE elementos más detallados de los registros clínicos requiere que el personal que administra estas 2 fuentes de datos distintas trabaje en colaboración sobre las definiciones de los datos o para combinar la extracción de datos electrónica con la extracción manual de los conceptos que no permitan el método automático.
- 3.
Generalmente, los registros disponen de procesos para asegurar la calidad de los datos; en cambio, los datos de las HCE no suelen estar sujetos a una auditoría formal.
Los principales obstáculos derivan de la necesidad de personal especializado, programas informáticos, sistemas de almacenamiento de datos y centros de análisis. La puesta en marcha de los registros a menudo se basa en líderes médicos voluntarios y comprometidos. Es crucial obtener el apoyo de las instituciones públicas (organismos gubernamentales, hospitales y otras instituciones de salud pública) o de la industria. Para ello, es de capital importancia considerar estos gastos como inversiones para el futuro.
La aplicación generalizada de registros requiere la estandarización de las definiciones de los datos en la totalidad de los distintos registros existentes en un campo concreto. Son precisas normas de calidad de los datos, pero no siempre se dispone de ellas o, si las hay, pueden no haberse aplicado de manera amplia y adecuada.
El corto tiempo para la obtención de los datos (solo el periodo de hospitalización) y el carácter específico de los resultados evaluados (solo muerte) conllevan limitaciones y deficiencias. A este respecto, la extracción de los datos a partir de las HCE podría facilitar el seguimiento longitudinal de los pacientes y la captación de datos de una amplia variedad de parámetros de valoración.
Los programas de auditoría regulares son esenciales para verificar la buena exactitud de los datos y que estén en consonancia con las normas de calidad actualmente aceptadas75,76.
Superación de los obstáculos y las limitaciones de los registrosEl empleo de métodos estandarizados es crucial para la calidad de los datos de los registros, a la vez que facilita las comparaciones entre las observaciones realizadas en diferentes registros. Esto es de capital importancia cuando hay un gran número de centros y países que participan en un mismo registro. En Europa, se han desarrollado las normas de calidad CARDS (Cardiology Audit and Registration Data Standards)77.
En Estados Unidos, un informe del grupo de trabajo sobre normas de calidad de datos (Data Standards Workgroup) del National Cardiovascular Research Infrastructure Project ha proporcionado unos datos cardiovasculares estandarizados para registros clínicos78.
En una época de extrema contención de los costes, es preciso resaltar el papel de los registros en la mejora de la asistencia clínica y el consiguiente beneficio económico. Cuando sea posible, debe haber una conexión entre los registros clínicos y las HCE, de manera que se minimice la entrada manual de datos y, por lo tanto, el esfuerzo y el coste requeridos. Los registros de nueva creación y los más maduros deben compartir sus datos y aprender unos de otros.
Los métodos de gestión de grandes cantidades de datos y los métodos estadísticos potentes y sofisticados serán útiles, sin duda alguna, para tener mayores posibilidades de extraer un conocimiento interpretable y válido a partir de los registros, superando en cierta medida las limitaciones que tienen los estudios no aleatorizados. Las técnicas de «aprendizaje automático» (machine learning/deep learning) y las redes neuronales (neural networks) pueden ser útiles para predecir la progresión de la enfermedad y los efectos específicos del tratamiento en pacientes individuales basándose en series de datos amplias, que complementen lo obtenido en otros niveles de investigación. El machine learning permite elaborar algoritmos que utilizan datos clínicos fácilmente disponibles para identificar a los pacientes con un trastorno especifico (enfermedad o estadio de la enfermedad)79–81.
Los registros clínicos deben adaptarse para obtener medidas del resultado centradas en el paciente. El extraordinario desarrollo de tecnologías de seguimiento automático con fines sanitarios brinda una gran oportunidad. El desarrollo de nuevos instrumentos electrónicos para recoger datos estandarizados de resultados presentados por los pacientes será de inestimable valor82,83.
Registros impulsados por los pacientesTradicionalmente, los investigadores son quienes han generado los registros. Los registros impulsados por los pacientes son similares en muchos aspectos a los generados por los investigadores, en cuanto a su definición, su finalidad y sus características, pero en los registros impulsados por los pacientes, estos y sus familiares «impulsan» el registro gestionando o controlando la recolección de datos, el programa de investigación para los datos o la difusión de la investigación.
En particular, los registros de pacientes generados por pacientes han sido criticados en varios aspectos y son necesarias mejoras en la estandarización y la calidad de los datos, la educación sanitaria de los pacientes para mejorar su capacidad de participación y aspectos relativos a la competencia entre los pacientes y los cuidadores.
Sin duda alguna, los registros y las redes de pacientes impulsados por los propios pacientes son un factor en rápida evolución que contribuye a la investigación y en especial a la investigación centrada en la mejora directa de la práctica clínica. Estas entidades difuminan las fronteras tradicionales y eliminan los obstáculos a la participación de los pacientes, las familias y los defensores del paciente y el control de la investigación, la traslación y la difusión de los resultados. Ha surgido un claro movimiento destinado a conectar las diversas organizaciones de pacientes y los registros de pacientes respecto a un único trastorno, para formar redes más amplias que unifiquen, estandaricen y optimicen la obtención de datos y los procesos de generación de la investigación84,85.
Euro Heart SurveysLa Sociedad Europea de Cardiología diseñó el programa Euro Heart Survey para evaluar la aplicabilidad de la medicina basada en la evidencia, la aplicación de guías en la práctica clínica y los resultados de diferentes estrategias de tratamiento de los pacientes85. Como ya se ha mencionado, las normas de calidad de los datos (CARDS) se han elaborado para fomentar la homogeneidad en la obtención de datos en todos los países de Europa77,86.
La experiencia inspiradora de SueciaEl registro cardiovascular SWEDEHEART se inició en 2009 mediante la fusión de 4 registros preexistentes, RIKS-HIA: registro de asistencia coronaria aguda (1995), SEPHIA: registro de prevención secundaria (2005), SCAAR: registro de angiografía e intervención (1998) y registro de cirugía cardiaca de Suecia (1992)87.
A pesar de su carácter no obligatorio, el registro cubre toda la actividad del país y está vinculado con registros obligatorios del Estado, como el registro nacional de pacientes, el registro de causas de muerte y el registro de medicamentos dispensados. Los coordinadores elaboran y presentan un registro anual de actividades88.
En palabras de sus propios líderes, las claves del éxito son las siguientes: a) creación por cardiólogos e impulso por entusiastas nacionales y locales; b) uso de un proceso de registro sencillo; c) usuarios muy motivados con acceso directo a los informes y las estadísticas del estudio; d) obtención de un beneficio inmediato en los informes online de la unidad local; e) posibilidad de una comparación abierta de los resultados de los hospitales; f) conceptualización del SCAAR no solo como un registro, sino como una herramienta que aporta una información clínica que influye en la calidad de la asistencia; g) flexibilidad y usuarios con capacidad de influir en el contenido, y h) un alto grado de transparencia.
Por lo que respecta a los métodos de investigación derivados, los aspectos funcionales que han sido cruciales son los siguientes:
- •
Cada hospital es propietario de sus propios datos; la participación en bases de datos científicas e informes de ámbito nacional es voluntaria.
- •
Los proyectos de investigación basados en bases de datos nacionales deben ser aprobados por el grupo directivo de SWEDEHEART.
- •
Todos los proyectos deben ser aprobados por un comité de ética.
- •
Toda base de datos se anonimiza antes de que llegue al científico.
- •
Los análisis estadísticos se realizan a menudo en colaboración con un epidemiólogo/bioestadístico de un centro nacional competente.
El registro ha dado origen a un gran número de publicaciones que abordan múltiples temas de la asistencia cardiovascular. Tan solo en 2015 se generaron 57 publicaciones a partir de él.
El registro afronta ahora avances recientes y futuros, como los siguientes: a) un módulo de asignación aleatoria para los ensayos prospectivos y aleatorizados basados en el registro; b) la integración con la HCE del paciente; c) la capacitación para la introducción en el sistema de la notificación directa procedente de los pacientes; d) la integración con módulos para la obtención de muestras de sangre para biobancos destinados a la investigación genética y proteómica, y e) colaboraciones internacionales: MINAP (Reino Unido), Infarctus Regiszter (Hungría) y ACTION (Estados Unidos).
Registros de los Grupos de Trabajo de la Sociedad Española de CardiologíaLos grupos de trabajo de la Sociedad Española de Cardiología han venido realizando y publicando sus registros de actividad con una periodicidad anual. Estos registros han permitido conocer las tendencias de la asistencia cardiovascular en las últimas 2 décadas y las diferencias regionales existentes respecto al uso de diferentes intervenciones cardiovasculares, como la intervención coronaria percutánea, los marcapasos, las ablaciones, los desfibriladores automáticos implantables o el trasplante de corazón. Sin embargo, no son registros de pacientes, en general no presentan parámetros de valoración clínicos y la fiabilidad de los datos se da por hecha sin que se hayan llevado a cabo auditorías o controles de calidad. El registro de Suecia deberá inspirar los avances en nuestro país como un objetivo que permitirá mejorar la calidad de la asistencia e incrementará el potencial de investigación89–93.
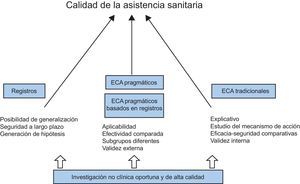
CONCLUSIONESSe ha revisado la investigación preclínica y clínica tal como se lleva a cabo actualmente, destacando las limitaciones de cada una, resaltando los obstáculos existentes y proponiendo posibles soluciones. Desde los estudios en animales hasta los modelos computacionales, desde los ECA tradicionales hasta los ECA pragmáticos basados en registros, desde los registros específicos basados en documentos en papel hasta los registros nacionales vinculados a las HCE, todo ello son avances que contribuyen a la generación de evidencia y, en última instancia, alcanzar una mejor calidad de la asistencia para todos (figura 3).

Parece absolutamente claro para todos que es necesaria una innovación continua y un cambio en el modo en que se realiza la investigación clínica. Para ello será preciso un fuerte compromiso de todos los profesionales de la salud, los investigadores, los políticos y la sociedad en general.
¡Esta es una llamada a la acción!
«Las ideas no duran mucho. Hay que hacer algo con ellas»
Santiago Ramón y Cajal
CONFLICTO DE INTERESESE.R. Edelman cuenta con una financiación parcial a través de una subvención de los National Institutes of Health (R01 GM 4903). Los demás autores no tienen conflictos de intereses que declarar.