
La amiloidosis cardiaca es una enfermedad infiltrativa por depósito extracelular de proteínas. De las proteínas proamiloidóticas a nivel cardiaco, la transtiretina produce una de las formas más frecuentes de amiloidosis cardiaca, bien por mutaciones o bien en su forma natural (wild-type) conocida previamente como amiloidosis senil. Hasta muy recientemente, el diagnóstico de amiloidosis por transtiretina (ATTR) se producía en reducidas ocasiones y requería confirmación histológica, por lo que establecer el diagnóstico constituía un verdadero reto en la práctica clínica habitual. Además, las opciones terapéuticas específicas para alterar el curso clínico de la enfermedad eran muy limitadas. Sin embargo, avances en el campo de la imagen cardiaca y en la estrategia diagnóstica de la enfermedad están facilitando un reconocimiento creciente de la ATTR. De forma adicional, diversos compuestos capaces de modificar la historia de la enfermedad se encuentran en fases finales de investigación, con resultados prometedores. Dado que una terapia efectiva parece estar cada vez más próxima, se hace imprescindible que los cardiólogos conozcan esta patología en profundidad y estén familiarizados con su diagnóstico y tratamiento. En esta revisión se repasará detalladamente el amplio espectro clínico de la ATTR, así como los recientes avances en el diagnóstico y tratamiento de esta entidad.
Palabras clave
La amiloidosis es una enfermedad de depósito, consecuencia del acúmulo extracelular de fibras que proceden de proteínas con una estructura inestable que se pliegan, agregan y terminan depositándose1. Secundariamente a ese depósito, se produce una alteración estructural que conlleva disfunción de distintos órganos y sistemas2.
Típicamente, las fibras de amiloide, insolubles y resistentes a la proteólisis, se tiñen con rojo Congo, proporcionando birrefringencia verde bajo luz polarizada3. De las más de 30 proteínas que pueden dar lugar a amiloide, solo 5 lo hacen de forma significativa a nivel cardiaco1:
- •
Cadenas ligeras, que dan lugar a amiloidosis primaria (AL).
- •
Transtiretina (TTR), que origina amiloidosis por TTR (ATTR).
- •
Apolipoproteína A.
- •
Fibrinógeno.
- •
Componente sérico A, que produce amiloidosis secundaria.
La mayoría de pacientes con amiloidosis cardiaca están afectados por AL o ATTR, siendo la forma AL la que tradicionalmente se ha considerado como más frecuente en países desarrollados3.
De hecho, la mayoría de la información sobre amiloidosis cardiaca estaba basada en AL. Sin embargo, mientras el número de pacientes con AL se mantiene estable, el número de pacientes diagnosticados con ATTR se ha incrementado en los últimos años y se ha llegado a considerar que probablemente la ATTR es mucho más prevalente que la AL2.
Tradicionalmente, la ATTR ha sido una entidad objeto de abundantes diagnósticos erróneos o bien de significativas demoras hasta su correcto diagnóstico. Los motivos han sido diversos y van desde la heterogeneidad en su presentación, la necesidad de demostración histológica o la escasez de equipos especializados hasta la consideración nihilista por parte de algunos cardiólogos de enfermedad rara y sin opciones de tratamiento2,3.
Sin embargo, estas concepciones están cambiando. El diagnóstico tiene implicaciones en cuanto al manejo de los pacientes y, dado el desarrollo de terapias específicas que pueden retrasar o estabilizar el depósito y que son más eficaces en fases iniciales, su realización de manera precoz es importante. En esta revisión se repasarán los avances en el diagnóstico y tratamiento de la ATTR, un campo con significativos progresos recientes y cargado de esperanza para los pacientes.
AMILOIDOSIS CARDIACA POR TRANSTIRETINALa TTR es una proteína plasmática, tetramérica, que se encarga de transportar tiroxina y la proteína ligada al retinol. Se sintetiza principalmente en el hígado y, en pequeña cuantía, en los plexos coroideos y en la retina4.
La TTR tiene predisposición a disgregarse en dímeros y monómeros, capaces de ensamblarse en fibras y depositarse. Las mutaciones puntuales o el efecto de la edad pueden incrementar esta predisposición, dando lugar a las 2 formas clínicas de la ATTR: la forma hereditaria (ATTRm) y la forma natural (ATTRwt).
AMILOIDOIDOSIS HEREDITARIA POR TRANSTIRETINAActualmente se conocen más de 120 mutaciones que pueden causar ATTRm. Se transmiten de forma autosómica dominante, con penetrancia variable4. Dada la gran diversidad geográfica, es difícil establecer su prevalencia; pero la ATTRm se considera una enfermedad rara, con una prevalencia inferior a 1 por cada 100.000 habitantes2 (tabla 1).
Principales características clínicas y diagnósticas de la amiloidosis cardiaca por transtiretina hereditara y senil
| ATTRwt | ATTRm | |
|---|---|---|
| Prevalencia | Desconocida. Parece muy frecuente | < 1:100.000 |
| Estudio genético | Ausencia de mutaciones en TTR | Mutación en TTR |
| Edad de presentación habitual | > 60 años | Variable según mutación causal |
| Sexo | Predominio masculino. 80% casos | Predominio masculino, con fenotipo más agresivo |
| Manifestaciones extracardiacas | • Síndrome del túnel del carpo (33-49%) • Estenosis del canal lumbar • Rotura atraumática del tendón bíceps braquial (32%) | • Polineuropatía sensitivo-motora bilateral ascendente • Disautonomía: hipotensión ortostática, diarrea-estreñimiento, disfunción eréctil • Afectación ocular: glaucoma, depósitos intravítreos, pupila festoneada |
| Afectación cardiaca | Constante | Variable según mutación causal |
| Presentación cardiaca | • Insuficiencia cardiaca (53-86%) • Trastornos de la conducción • FA (43-67%) • EAo degenerativa | • Trastornos de conducción • Insuficiencia cardiaca • FA poco frecuente (10%) |
| Técnicas diagnósticas | ||
| ECG | • Patrón de seudoinfarto (63-66%) • Bajo voltaje (22-33%) • HVI Sokolow (6-13%) | • Patrón de seudoinfarto (18-69%) • Bajo voltaje (2-25%) • HVI Sokolow (3-8%) |
| ECO | • Hipertrofia moderada-grave • FEVI leve-moderadamente deprimida (30%) | • Hipertrofia moderada • FEVI habitualmente conservada |
| Resonancia magnética cardiaca | • Realce tardío • Elevación de T1 nativo y VEC | |
| Gammagrafía 99mTc-DPD | • Grado 2-3 | • Grado 0: portadores asintomáticos • Grado 1: afección cardiaca inicial • Grado 2-3: afección cardiaca significativa |
ATTRm: amiloidosis hereditaria por transtiretina; ATTRwt: amiloidosis natural; EAo: estenosis aórtica; ECG: electrocardiograma; ECO: ecocardiograma; FA: fibrilación auricular; HVI: hipertrofia ventricular izquierda; FEVI: fracción de eyección del ventrículo izquierdo; TTR: transtiretina; VEC: volumen extracelular.
Dado que las primeras mutaciones en TTR se reportaron como casos de polineuropatía amiloide familiar (o enfermedad de Andrade), la ATTRm se ha considerado hasta hace poco una enfermedad fundamentalmente neurológica. Sin embargo, datos recientes muestran que el corazón está afectado en más de la mitad de los casos3.
Hay una fuerte correlación genotipo-fenotipo, con mutaciones que producen un cuadro predominantemente neurológico y otras un cuadro fundamentalmente cardiaco3. Sin embargo, la dicotomización en ATTRm de predominio cardiaco o neurológico puede ser una mera simplificación y el espectro de la enfermedad es mucho más continuo entre ambas formas clínicas.
La mutación Val30Met (Val50Met según la nomenclatura actualizada que añadió 20 posiciones a la denominación tradicional de mutaciones en ATTRm) es la mutación más frecuente a nivel mundial y es endémica en Portugal, Japón y Suecia. En Portugal, la incidencia estimada es de 1 de cada 538 individuos2. En España, la isla de Mallorca y la localidad de Valverde del Camino (Huelva) se consideran también zonas endémicas de ATTRm. La prevalencia estimada en Mallorca, considerando solo a los pacientes sintomáticos, es de 3 de cada 100.000 habitantes5.
La mutación Val30Met da lugar a afección predominantemente neurológica con una polineuropatía simétrica, sensitivomotora, ascendente y de inicio en miembros inferiores. Puede asociar disautonomía con hipotensión ortostática, disfunción eréctil, incontinencia urinaria y síntomas gastrointestinales. Se inicia generalmente al final de la segunda o tercera década de la vida y hasta el 43% de los portadores presentan una afección cardiaca que es causa frecuente de mortalidad4 (tabla 1).
Destaca por su relevancia la mutación Val122Ile (p. Val142Ile), que está presente en un 3-4% de la población norteamericana de raza negra3. Aunque su penetrancia es incompleta3, esta mutación se ha asociado a un 47% de aumento de riesgo de desarrollar insuficiencia cardiaca (IC)6. Recientemente, un estudio señaló la amiloidosis Val122Ile como la cuarta causa de IC en población afrocaribeña británica7. Aunque hasta el 30% de los pacientes con esta mutación pueden presentar datos de neuropatía leve6, el fenotipo clínico es habitualmente similar a la ATTRwt4. Val122Ile no debe considerarse una mutación exclusiva de la población de raza negra, ya que también puede estar presente en población caucásica y, por poner un ejemplo, nosotros la hemos identificado en 4 familias españolas de raza blanca sin antepasados de raza negra.
AMILOIDOSIS POR TRANSTIRETINA NATURALLa ATTRwt fue descrita por primera vez en 1876. Anteriormente se la denominaba amiloidosis senil, pero su diagnóstico en sujetos en la sexta y séptima décadas de la vida ha hecho que este término esté en desuso. Como curiosidad, el caso más precoz reportado hasta la fecha corresponde a un sujeto estadounidense de 47 años de edad8.
La prevalencia de la ATTRwt no se conoce con exactitud. Sin embargo, hay varios datos que sugieren que es una entidad infradiagnosticada y que probablemente se trate la amiloidosis cardiaca más frecuente2,3. Los datos que apoyan esta hipótesis son:
- •
La prevalencia de depósito de TTR en autopsias realizadas a personas > 80 años es del 25%3.
- •
En autopsias de pacientes con IC con fracción de eyección preservada (ICFEp), el 5% presentaba un depósito moderado-grave de TTR9.
- •
Recientemente, nuestro grupo encontró una prevalencia del 13% en pacientes > 60 años ingresados por ICFEp y con hipertrofia ventricular izquierda (HVI) ≥ 12mm10.
A diferencia de la ATTRm, la ATTRwt es una enfermedad esporádica con inicio típico a partir de los 70 años4. Clásicamente, ha destacado su predominio en varones y constituyen hasta el 89-98% de las series publicadas11,12. Sin embargo, nuestro grupo ha descrito recientemente que el 20% de la serie de pacientes con ATTRwt diagnosticados en nuestro centro y en Bolonia son mujeres y otros estudios autópsicos también sugieren que la enfermedad en las mujeres puede ser mayor de lo que se ha publicado; por lo que el sexo femenino no debería desviar la sospecha clínica de ATTRwt13 (tabla 1).
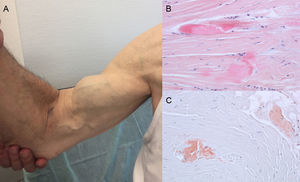
Los datos de las autopsias muestran que el depósito de TTR en la ATTRwt se produce de forma dispersa en distintos órganos. Sin embargo, el tropismo cardiaco de la TTR hace que el depósito sea mucho mayor en este órgano y que su afectación sea la manifestación clínica principal4. Los pacientes pueden presentar síntomas derivados del depósito de TTR a nivel extracardiaco como estenosis del canal lumbar, rotura atraumática del tendón del bíceps o «signo de Popeye» y síndrome del túnel del carpo (STC)3 (figura 1). Todos estos signos ayudan a sospechar el diagnóstico y establecerlo con prontitud. Aunque el STC puede acompañar a otros subtipos de amiloidosis, en la ATTRwt lo hace con mayor frecuencia. Típicamente, el depósito puede preceder a las manifestaciones cardiacas en varios años6 y puede usarse como signo indicativo en pacientes ancianos con HVI, especialmente si el STC es bilateral, no asociado a actividades laborales específicas y en presencia de clase funcional de la New York Heart Association ≥ II (datos no publicados).
. B y C: tinción con hematoxilina-eosina (B) y con rojo congo (C), ambas ×200, de muestra de ligamento del carpo que muestra haces de colágeno denso con presencia de material acelular. Cortesía de la Dra. Clara Salas Antón.")
Signos y síntomas en amiloidosis por transtiretina. A: rotura atraumática del tendón bíceps derecho («signo de Popeye»). B y C: tinción con hematoxilina-eosina (B) y con rojo congo (C), ambas ×200, de muestra de ligamento del carpo que muestra haces de colágeno denso con presencia de material acelular. Cortesía de la Dra. Clara Salas Antón.
El amiloide puede infiltrar cualquier estructura cardiaca1. Típicamente, el depósito produce un engrosamiento de las paredes ventriculares que progresivamente empeora la distensibilidad dando lugar a una disfunción diastólica grave. Por ello, la ATTR se ha incluido tradicionalmente como una de las causas de miocardiopatía restrictiva.
Sin embargo, el espectro clínico de presentación de la ATTR es mucho más amplio y heterogéneo. La IC es la forma más frecuente de presentación de ATTR. Tal como se ha comentado previamente, nuestro grupo publicó en 2015 que un protocolo basado en el empleo de 99mTc-3,3-difosforo-1,2-ácido propanodicarboxílico (Tc-DPD) permitía diagnosticar ATTRwt en una proporción significativa (13%) de los pacientes mayores de 60 años ingresados por ICFEp10. A partir de este trabajo, la gammagrafía con 99mTc-DPD se ha incluido en las guías europeas de IC como herramienta útil para la identificación de pacientes con ATTR14. Sin embargo, la sospecha de ATTR no debe restringirse a ICFEp ya que, a medida que el depósito amiloideo progresa, la función contráctil empeora; por lo que la ATTR puede cursar con distintos grados de disfunción sistólica.
La ATTR es una fenocopia de miocardiopatía hipertrófica (MCH) y puede confundirse con ella. En un reciente estudio multicéntrico francés se ha comunicado que el 5% de los pacientes con MCH presentan ATTRm15. No obstante, nuestra experiencia no es acorde con estos resultados y es posible que la alta tasa registrada pudiese estar en relación con la alta tasa de población de raza negra presente en el país vecino.
Las alteraciones en la conducción cardiaca pueden ser la primera manifestación de la ATTR. La infiltración amiloide del sistema de conducción a nivel del nodo sinusal y aurículoventricular1 puede hacer necesaria la implantación de marcapasos (tabla 1). En un trabajo reciente de nuestro grupo (en combinación con el grupo de Bolonia) se constató que el 7% de los pacientes con ATTRwt comenzaron con trastornos de conducción13.
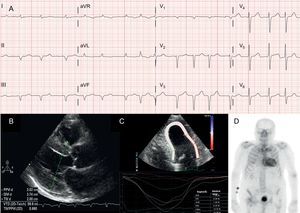
Las arritmias auriculares también son muy frecuentes en pacientes con ATTRwt13 (figura 2A) y la primera manifestación de la enfermedad puede ser incluso un accidente cerebrovascular4. De hecho, el grupo de la Clínica Mayo ha planteado recientemente que quizás debería descartarse la presencia de ATTRwt ante el diagnóstico de fibrilación auricular (FA) no valvular en ancianos8. En contraposición, la FA aparece en mucha menor medida en los pacientes con ATTRm (tabla 1).
. A: electrocardiograma de un paciente con amiloidosis por transtiretina, forma natural (ATTRwt), que muestra fibrilación auricular y patrón de seudoinfarto en derivaciones inferiores. B: ecocardiograma de paciente con amiloidosis por transtiretina hereditaria por mutación en Val30Met en el que se puede observar hipertrofia ventricular izquierda concéntrica marcada y leve derrame pericárdico. C: strain longitudinal regional de paciente con ATTRwt que muestra valores conservados en ápex, con reducción a nivel basal y medio. D: gammagrafía 99mTc-DPD (99mTc-ácido 3,3-difosfono-1,2-propanodicarboxílico) de paciente con ATTRwt que muestra captación biventricular y superior al hueso, correspondiente con grado 3 de Perugini.")
Técnicas diagnósticas en la amiloidosis cardiaca por transtiretina (ATTR). A: electrocardiograma de un paciente con amiloidosis por transtiretina, forma natural (ATTRwt), que muestra fibrilación auricular y patrón de seudoinfarto en derivaciones inferiores. B: ecocardiograma de paciente con amiloidosis por transtiretina hereditaria por mutación en Val30Met en el que se puede observar hipertrofia ventricular izquierda concéntrica marcada y leve derrame pericárdico. C: strain longitudinal regional de paciente con ATTRwt que muestra valores conservados en ápex, con reducción a nivel basal y medio. D: gammagrafía 99mTc-DPD (99mTc-ácido 3,3-difosfono-1,2-propanodicarboxílico) de paciente con ATTRwt que muestra captación biventricular y superior al hueso, correspondiente con grado 3 de Perugini.
Por último, resaltar la posibilidad de coexistencia de ATTR junto con una estenosis aórtica degenerativa. En el último año esta posibilidad se ha destacado en varios trabajos, y un estudio prospectivo ha comunicado una prevalencia de ATTRwt del 6% entre pacientes mayores de 65 años sometidos a reemplazo valvular aórtico16. En dicho estudio, los pacientes que presentaban ambas entidades mostraron un pronóstico posoperatorio mucho más desfavorable con respecto a los que no tenían ATTRwt (mortalidad del 50 frente al 6,9% tras una mediana de seguimiento de 2,3 años)16. Al mismo tiempo, otro estudio con Tc-DPD identificó a 5 pacientes con ATTRwt entre los 43 que presentaban estenosis aórtica de bajo flujo-bajo gradiente; lo que supone un 12% de prevalencia17. Indudablemente, la estenosis aortica grave y la ATTRwt comparten un mismo perfil demográfico de pacientes y queda por establecer cuál es el tratamiento más adecuado para los individuos con ambas patologías.
Utilidad de las técnicas diagnósticasEl diagnóstico de ATTR constituye un reto en la práctica clínica diaria. Aunque tanto el electrocardiograma como el ecocardiograma desempeñan un papel en el diagnóstico, nuevas técnicas no invasivas han adquirido un papel crucial en la evaluación de estos pacientes.
ElectrocardiogramaClásicamente, se ha considerado como dogma la asociación entre bajo voltaje y amiloidosis cardiaca3. En la práctica clínica, el criterio más ampliamente utilizado es una amplitud de QRS < 1mV en todas las derivaciones precordiales o < 0,5mV en todas las de los miembros1. Aunque la presencia de bajo voltaje electrocardiográfico en el contexto de HVI debe establecer la sospecha, la prevalencia en series contemporáneas de ATTR es tan baja como del 20-25%3,4,13. Además, la prevalencia varía según el criterio empleado. Así, el criterio de Sokolow (suma de onda S en V1 y onda R en V5 o V6 ≤ 1,5mV) puede incrementar la prevalencia hasta un 46-58%13. Para una mejor evaluación de la desproporción electrocardiograma/ecocardiograma, se promueve el empleo de la ratio entre el grosor ventricular izquierdo y el voltaje total de QRS2,3. Contrariamente, conviene saber que hasta un 20% de los pacientes con ATTR pueden cumplir criterios electrocardiográficos de HVI2,3.
Sin embargo, el hallazgo electrocardiográfico más frecuente en la mayor parte de las series de amiloidosis cardiaca es el patrón de seudoinfarto2,3,13 (figura 2A). Dada la posible afección del sistema de conducción, los bloqueos de rama completos o incompletos son también habituales3.
EcocardiogramaAunque el ecocardiograma constituye la piedra angular del diagnóstico inicial de ATTR, ningún hallazgo es específico3. Típicamente, la ATTR se ha asociado con un ventrículo izquierdo normal o pequeño, con hipertrofia concéntrica3. El consenso del décimo congreso internacional de amiloidosis de 2004 estableció como criterio ecocardiográfico de afección cardiaca por AL la presencia de HVI con un punto de corte de 12mm a nivel septal, en ausencia de otras causas de HVI4. Este criterio se extrapoló posteriormente al resto de formas de amiloidosis (figura 2B), lo que confirió un alto grado de especificidad pero baja sensibilidad.
Aunque clásicamente se ha descrito la HVI concéntrica, datos de series actuales han señalado que alrededor del 20% presenta HVI asimétrica13.
A pesar de la asociación clásica de fracción de eyección del ventrículo izquierdo (FEVI) normal o levemente disminuida2, el rango de FEVI es muy variable8. En un estudio reciente de ATTRwt de la Clínica Mayo, casi la mitad de los pacientes presentaba una FEVI < 50%8 y en nuestra serie ocurría en el 37%13. Además, la evaluación de la función sistólica mediante FEVI en la amiloidosis cardiaca tiene limitaciones; pues valores ligeramente deprimidos implican ya afección cardiaca relevante. Las velocidades obtenidas mediante Doppler tisular, la deformación o strain y la fracción de contracción miocárdica permiten superar esas limitaciones y se han propuesto como índices más adecuados para la evaluación de la función cardiaca2.
Otros signos ecocardiográficos clásicos son la hipertrofia de ventrículo derecho, la dilatación biauricular, el derrame pericárdico leve, el engrosamiento de las válvulas auriculoventriculares y el del septo interauricular; así como el aspecto granular o sparkling del miocardio3,6. Sin embargo, algunos de estos hallazgos provienen de series muy seleccionadas de pacientes en fases avanzadas de enfermedad, por lo que no se debe esperar encontrar todas estas características para establecer la sospecha1.
El strain regional es una técnica muy útil para el diagnóstico precoz de los pacientes con ATTR. En los pacientes con ATTR el strain longitudinal está disminuido en los segmentos basales y medios y conservado en segmentos apicales18 (figura 2C). Ese patrón típico ayuda a obtener el diagnóstico diferencial con otras cardiopatías4.
BiomarcadoresA diferencia de lo que ocurre en la AL, hay menos datos sobre el papel de la fracción aminoterminal del propéptido natriurético cerebral (NT-proBNP) y la troponina en la ATTR4. Las cifras de NT-proBNP en la ATTR tienden a ser más bajas respecto a la AL4, pudiendo reflejar 2 mecanismos fisiopatológicos distintos: toxicidad directa de las cadenas ligeras en la AL frente a daño inducido de los protofilamentos en la ATTR.
Recientemente, el grupo de la clínica Mayo ha propuesto un sistema de estratificación similar al vigente para la AL. En una cohorte de 360 pacientes con ATTRwt ambos biomarcadores demostraron ser predictores de mortalidad. Los pacientes en estadio III (NT-proBNP > 3.000 pg/ml y troponina T > 0,05 ng/ml) presentaban una mediana de supervivencia de 20 meses respecto a los 66 y 40 meses de los pacientes en estadios I y II (ninguno o solo un marcador por encima de los puntos de corte establecidos, respectivamente)8.
Resonancia magnética cardiacaLa resonancia magnética cardiaca (RMC) proporciona información estructural y funcional y permite caracterizar la composición tisular del miocardio3. Resulta fundamental en la identificación precoz de la ATTR, así como en el diagnóstico diferencial con otras cardiopatías.
La caracterización tisular mediante RMC se basa en:
- •
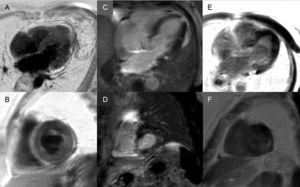
Presencia de realce tardío: un patrón subendocárdico global es virtualmente patognomónico de amiloidosis cardiaca, pero solo está presente en alrededor de una cuarta parte de los pacientes y son también compatibles otros patrones como el transmural (el más frecuente) o el parcheado (figura 3). A pesar de su alta sensibilidad y especificidad, se debe tener en cuenta la posibilidad de ausencia de realce tardío (15% de los casos) y, en nuestra experiencia, un porcentaje no desdeñable de falsos negativos por razones técnicas3. Se ha descrito que el patrón de realce transmural conlleva un peor pronóstico evolutivo y es un predictor independiente de mortalidad19.
 que muestran depósito patológico de gadolinio difuso transmural. C y D: secuencias de realce tardío en plano de 4 cámaras y eje corto a nivel basal, respectivamente, de paciente con ATTRm que muestran depósito patológico de gadolinio parcheado, con área focal inferoseptal inferior e inferolateral basal. E y F: secuencias de realce tardío en plano de 4 cámaras y eje corto a nivel apical, respectivamente, de paciente con ATTRm que muestran depósito patológico extenso transmural, respetando únicamente segmentos anterolaterales basal y medios. Cortesía del Dr. Jesús González Mirelis.") Figura 3.
Figura 3.Diversidad de patrones de realce tardío por resonancia magnética cardiaca en amiloidosis por transtiretina. A y B: secuencias de realce tardío en plano de 4 cámaras y eje corto a nivel medial, respectivamente, de paciente con amiloidosis por transtiretina hereditaria (ATTRm) que muestran depósito patológico de gadolinio difuso transmural. C y D: secuencias de realce tardío en plano de 4 cámaras y eje corto a nivel basal, respectivamente, de paciente con ATTRm que muestran depósito patológico de gadolinio parcheado, con área focal inferoseptal inferior e inferolateral basal. E y F: secuencias de realce tardío en plano de 4 cámaras y eje corto a nivel apical, respectivamente, de paciente con ATTRm que muestran depósito patológico extenso transmural, respetando únicamente segmentos anterolaterales basal y medios. Cortesía del Dr. Jesús González Mirelis.
(0.15MB). - •
Valores elevados de T1: el mapeo T1 es una técnica en la cual una señal cuantitativa del miocardio se mide antes (T1 nativo) o después de la administración de contraste. El T1 nativo está muy elevado en la amiloidosis cardiaca3. Dado que no requiere la administración de contraste, es una técnica que se puede emplear en insuficiencia renal; pudiendo ser incluso anormal antes de que se observe HVI3. Los valores de T1 nativo son más elevados en ATTR que en MCH y controles (1.097 ± 43ms frente a 1.026 ± 64ms y frente a 9,67 ± 34ms, respectivamente; p < 0,0001), pero menores que en la AL (1.130 ± 68ms; p = 0,01)20.
- •
El cálculo del volumen extracelular (VEC) tras la administración de contraste permite evaluar el incremento del espacio extracelular. Los valores del VEC en la amiloidosis cardiaca son más elevados en comparación con otras cardiopatías, a excepción de las zonas de infarto21. Nuestro grupo, en colaboración con otros centros nacionales, comunicó en 2016 cómo el VEC permite identificar la afección cardiaca en la ATTRm y lo correlacionó por primera vez con el grado de deterioro neurológico, apoyando el empleo de esta técnica en el diagnóstico precoz y seguimiento de la ATTRm22.
 que muestran depósito patológico de gadolinio difuso transmural. C y D: secuencias de realce tardío en plano de 4 cámaras y eje corto a nivel basal, respectivamente, de paciente con ATTRm que muestran depósito patológico de gadolinio parcheado, con área focal inferoseptal inferior e inferolateral basal. E y F: secuencias de realce tardío en plano de 4 cámaras y eje corto a nivel apical, respectivamente, de paciente con ATTRm que muestran depósito patológico extenso transmural, respetando únicamente segmentos anterolaterales basal y medios. Cortesía del Dr. Jesús González Mirelis.")
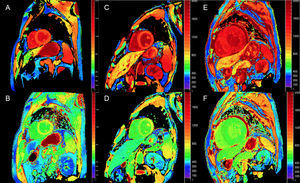
En tanto que son cuantitativas, las técnicas de mapeo T1 y de cálculo del VEC pueden ser de gran utilidad para el diagnóstico precoz, el seguimiento clínico y la evaluación de la respuesta al tratamiento en la ATTR (figura 4).
 en resonancia magnética cardiaca 3T de control sano, paciente con amiloidosis por transtiretina y amiloidosis primaria por cadenas ligeras. A y B: mapa T1 nativo y volumen extracelular (VEC), respectivamente, en control sano, que muestra valores normales (VEC = 0,214). C y D: mapa T1 nativo y VEC, respectivamente, en paciente con amiloidosis por transtiretina hereditaria con daño neurológico y afección cardiaca incipiente: elevación discreta de T1 nativo y VEC levemente elevado de 0,361. E y F: mapa T1 nativo y VEC, respectivamente, en paciente con amiloidosis cardiaca por transtiretina forma natural: elevación de T1 nativo y VEC muy elevado de 0,626, que refleja una infiltración amiloide masiva. Cortesía del Dr. Jesús González-Mirelis. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Mapas T1, antes y después de contraste, con inversión-recuperación de look-locker modificada (MOLLI) en resonancia magnética cardiaca 3T de control sano, paciente con amiloidosis por transtiretina y amiloidosis primaria por cadenas ligeras. A y B: mapa T1 nativo y volumen extracelular (VEC), respectivamente, en control sano, que muestra valores normales (VEC = 0,214). C y D: mapa T1 nativo y VEC, respectivamente, en paciente con amiloidosis por transtiretina hereditaria con daño neurológico y afección cardiaca incipiente: elevación discreta de T1 nativo y VEC levemente elevado de 0,361. E y F: mapa T1 nativo y VEC, respectivamente, en paciente con amiloidosis cardiaca por transtiretina forma natural: elevación de T1 nativo y VEC muy elevado de 0,626, que refleja una infiltración amiloide masiva. Cortesía del Dr. Jesús González-Mirelis. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
En los años ochenta del pasado siglo, la observación de captación cardiaca de varios trazadores de difosfonato de hueso se correlacionó histológicamente con la presencia de amiloide a nivel cardiaco23. El mecanismo por el que se produce dicha captación no se conoce con exactitud, pero se especula que está relacionado con el contenido de calcio de los depósitos amiloideos.
Un primer estudio del grupo de Bolonia con 99mTc-DPD reportó la captación a nivel cardiaco en 15 pacientes con ATTR y su ausencia en 10 con AL mediante la aplicación de una puntuación basada en la captación biventricular igual o superior respecto al hueso (puntuación de Perugini)24 (figura 2D). Otros grupos, entre ellos el nuestro, comunicó posteriormente hallazgos similares25. Cabe destacar que una captación leve (puntuación 1) y una captación moderada (puntuación 2) puede encontrarse en un 30 y un 10%, respectivamente, de los pacientes con AL24.
Dada su alta sensibilidad y especificidad, esta técnica es de enorme utilidad para establecer el diagnóstico de ATTR y puede mostrar afección cardiaca cuando la ecocardiografía y la RMC todavía son normales. De hecho, no son raros los casos diagnosticados de forma incidental a raíz de una gammagrafía realizada por indicaciones oncológicas o reumatológicas26.
El Tc-DPD no está disponible en Estados Unidos, pero se han descrito resultados similares empleando 99mTc-PYP (pirofosfato)27.
Otros radiotrazadores se encuentran en estudio en la actualidad. En este sentido, 18F-florbetapir, ya aprobado para la imagen de beta-amiloide cerebral4, ha sido estudiado en pacientes con AL y ATTR. Los resultados demostraron que 18F-florbetapir detecta los depósitos amiloideos cardiacos AL y ATTR28. Aunque los datos disponibles provienen solo de casos29 y el alto coste del radiotrazador limita su uso, varios estudios están en marcha con la potencial ventaja con respecto al Tc-DPD de poder emplearse como técnica de despistaje para los dos tipos más frecuentes de amiloidosis.
Diagnóstico invasivoEl diagnóstico definitivo de ATTR se basa en la demostración histológica de fibras de amiloide. A pesar de existir depósitos extracardiacos, no todos los órganos tienen la misma probabilidad de mostrar amiloide por histología2. Hay pocos estudios acerca de la rentabilidad de la biopsia extracardiaca (grasa abdominal, gingival, glándula salival, gastrointestinal, etc.) en la ATTR, que es mayor en la ATTRm que en la ATTRwt. En cualquier caso, conviene destacar que una biopsia negativa de un órgano no afectado clínicamente no excluye el diagnóstico4.
La biopsia endomiocárdica está indicada en aquellos casos en los que no se consigue demostración extracardiaca o en los que la cardiopatía es la única afección, como ocurre en ATTRwt3,4. El riesgo es bajo (sobre todo en centros experimentados) y el error de muestreo es poco probable6.
Una vez confirmado el diagnóstico histológico de amiloidosis, que en ocasiones puede requerir la interpretación por parte de personal entrenado6, es crucial establecer el subtipo4. Para su clasificación, en la actualidad se dispone de una combinación de inmunohistoquímica, análisis genético y proteómica:
- •
La inmunohistoquímica se basa en el uso de anticuerpos específicos contra proteínas amiloides conocidas. Aunque los resultados suelen ser definitivos para TTR, la técnica es menos sensible en el reconocimiento de cadenas ligeras4.
- •
Para superar esta limitación, la espectrometría de masas proporciona resultados definitivos y es el método de referencia en el diagnóstico del subtipo de amiloide2. Aunque solo está disponible en centros expertos, resulta especialmente útil en casos dudosos o con positividad para varios anticuerpos en inmunohistoquímica, lo que en nuestra experiencia llega a ocurrir en alrededor del 20-30% de los casos4.
- •
Por último, puesto que no es posible distinguir desde un punto de vista clínico ni histológico la forma hereditaria (ATTRm) de la adquirida (ATTRwt), el estudio genético está recomendado en todos los casos de ATTR. La constatación de una mutación causal permite ofrecer consejo genético y la monitorización de portadores asintomáticos4,30, que podrían beneficiarse en el futuro de terapias que retrasen o incluso prevengan el inicio de la enfermedad31.
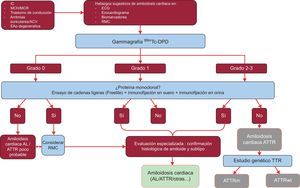
Hasta hace poco, el diagnóstico histológico de la ATTR era esencial3. Sin embargo, con el objetivo de facilitar el diagnóstico, en 2016 se publicó un trabajo multicéntrico internacional que proponía un nuevo algoritmo para el diagnóstico no invasivo de la ATTR32.
Se analizaron datos de 1.217 pacientes. La presencia de signos clásicos de amiloidosis cardiaca por técnicas de imagen, un grado 2-3 de captación en gammagrafía Tc-DPD/PYP, junto con la exclusión de una proteína monoclonal, confiere una sensibilidad y un valor predictivo positivo del 100% para el diagnóstico de ATTR32 (figura 5).

Algoritmo diagnóstico en pacientes con sospecha de amiloidosis cardiaca. Sistema de graduación de gammagrafía 99mTc-DPD: grado 0, ausencia de captación cardiaca; grado 1, captación leve menor que hueso; grado 2, captación moderada igual al hueso; grado 3, captación grave superior al hueso. ACV: accidente cerebrovascular; AL: amiloidosis primaria por cadenas ligeras; ATTR: amiloidosis por transtiretina; ATTRm: amiloidosis por transtiretina hereditaria; ATTRwt: amiloidosis senil; EAo: estenosis aórtica; ECG: electrocardiograma; IC: insuficiencia cardiaca; MCH: miocardiopatía hipertrófica; MCR: miocardiopatía restrictiva; RMC: resonancia magnética cardiaca; TTR: transtiretina.
Una parte fundamental de este algoritmo es la exclusión de proteína monoclonal que pudiese causar AL mediante el ensayo de cadenas ligeras libres en suero (Freelite) y la inmunofijación en sangre y orina. La presencia de proteína monoclonal requiere la realización de biopsia endomiocárdica para distinguir entre la ATTR y la AL32. Hasta un 5% de la población mayor de 65 años presenta una gammapatía monoclonal de significado incierto2. En ancianos, un incremento moderado de cadenas ligeras circulantes no debe llevar directamente al diagnóstico de AL. En centros de referencia se ha comunicado que hasta un 10% de los ancianos que presentaban ATTRwt y gammapatía monoclonal de significado incierto habían sido previamente mal diagnosticados de AL3,33. Es necesario realizar un correcto diagnóstico para evitar un uso inapropiado de la quimioterapia. Como curiosidad, en nuestro centro se han documentado 2 casos de pacientes con mieloma múltiple y ATTRwt concomitante demostrada por espectrometría de masas.
TRATAMIENTO DE LA AMILOIDOSIS CARDIACA POR TRANSTIRETINAEl tratamiento de los pacientes con ATTR tiene 2 objetivos: proporcionar soporte médico y detener o retrasar, si es posible, el depósito de amiloide mediante tratamiento específico.
Tratamiento médicoEl tratamiento de soporte de la afección cardiaca en la ATTR debe cubrir los aspectos que se describen a continuación.
Manejo de la insuficiencia cardiacaEl mantenimiento de un estado de euvolemia es fundamental en pacientes con amiloidosis cardiaca. Las medidas higienicodietéticas son muy importantes. Los diuréticos son la pieza clave en el tratamiento de la IC en la ATTR. Sin embargo —dado que un exceso diurético puede producir hipotensión y empeorar la situación clínica, sobre todo en la ATTRm, al asociar frecuentemente disfunción autonómica— el manejo debe de ser extremadamente cuidadoso.
En el tratamiento de la IC en la ATTR se debe tener en cuenta que la disfunción diastólica, y la reducción del volumen latido, condicionan una taquicardia compensadora que mantiene el gasto cardiaco. Por ello, el empleo de bloqueadores beta debe realizarse de forma cuidadosa e individualizada y la práctica habitual es su retirada en ausencia de problemas de control de frecuencia. En la ATTRwt esto es más importante, si cabe, por la frecuente presencia de trastornos de conducción6. Los antagonistas del calcio y la digoxina están contraindicados en la ATTR, dada la toxicidad por su potencial unión a las fibras de amiloide, incluso a dosis terapéuticas6.
A diferencia de lo que ocurre en la IC con disfunción sistólica por otras etiologías, no hay evidencia que apoye un beneficio pronóstico derivado del uso de bloqueadores beta, inhibidores de la enzima de conversión de la angiotensina o antagonistas de los receptores de la angiotensina II en la amiloidosis cardiaca. De hecho, su uso puede producir empeoramiento clínico por hipotensión y bajo gasto; en comunicaciones recientes se indica peor pronóstico en la ATTRm y efecto neutro en la ATTRwt34.
Manejo de arritmias auricularesEl manejo de la FA en la ATTR constituye un reto. El mantenimiento del ritmo sinusal a largo plazo es difícil. Sin embargo, un intento de cardioversión eléctrica puede ser razonable por la potencial mejoría clínica de los pacientes.
El riesgo tromboembólico de los pacientes con ATTR es muy elevado. Además, la infiltración crónica por amiloide puede condicionar una disfunción auricular mecánica que podría explicar por qué algunos pacientes sin FA desarrollan trombos auriculares. La decisión de indicar anticoagulación en ATTR no se debe basar en la puntuación CHADS2-VASc y debe ser la norma en caso de FA. Los eventos hemorrágicos son menos comunes que en la AL; por lo que en algunos centros se recomienda la anticoagulación en ritmo sinusal si hay una mala función auricular según velocidades de Doppler transmitral. En nuestro centro se han empleado anticoagulantes orales directos a algunos pacientes, pero no hay estudios comparativos acerca de su efectividad con respecto a los antagonistas de la vitamina K.
Papel de los dispositivosActualmente, las indicaciones para el implante de marcapasos en ATTR siguen las mismas recomendaciones que en la población general. Sin embargo, algunos grupos son partidarios del implante profiláctico, especialmente en pacientes con ATTRm en presencia de trastornos de conducción35. Nosotros no somos partidarios de esta estrategia preventiva y no hemos evidenciado una tasa tan elevada de trastornos de conducción que hiciese necesario una estrategia de implante de marcapasos profiláctico en sujetos con ATTRm.
El papel del desfibrilador automático implantable (DAI) en ATTR no está bien establecido. En series reducidas, el implante de DAI no mejoró la supervivencia de forma relevante; aunque el DAI actuó de forma apropiada en múltiples casos en los 2 primeros años36.
Trasplante cardiacoEl trasplante cardiaco ha desempeñado un papel minoritario en la ATTR debido a la naturaleza multiorgánica de la ATTRm y a la edad avanzada en la ATTRwt. Sin embargo, la ausencia de afección extracardiaca en la ATTRwt hace que sean buenos candidatos y en la bibliografía hay ejemplos de trasplantes en pacientes con ATTRwt menores de 70 años o con ATTRm y afección predominante cardiaca, con buenos resultados4.
Tratamiento específico de la amiloidosis cardiaca por transtiretinaEn el momento actual no hay ninguna terapia aprobada para el tratamiento específico de la ATTR cardiaca, aunque el trasplante hepático (TxH), aislado o en combinación con el cardiaco, ha sido empleado en ATTRm desde la década de 1990 como forma de eliminar la principal fuente precursora de TTR4.
Trasplante hepáticoSegún el registro mundial de trasplante de ATTRm37, más de 2.000 pacientes se han sometido a TxH en 20 países4. La supervivencia reportada en pacientes con mutación Val30Met y clínica predominantemente neurológica es superior al 50% a los 20 años3. Estos buenos resultados se basan en una estricta selección de pacientes según la edad, la mutación y el estadio de la enfermedad. Por ello, la indicación más aceptada es la de pacientes jóvenes, con mutación Val30Met y en fases iniciales de la enfermedad.
Sin embargo, las mayores limitaciones de esta técnica se basan en la escasez de donantes, la necesidad de inmunosupresión crónica, la edad avanzada en el momento de presentación y los peores resultados obtenidos en pacientes con mutaciones no-Val30Met.
Además, la teórica supresión de la producción de la proteína mutada se ve contrarrestada por un depósito progresivo de TTR nativa tras el trasplante4,6 cuyo mecanismo no se conoce con exactitud. De hecho, este depósito a nivel cardiaco condiciona la morbimortalidad tras el TxH.
Una mejor comprensión de la patogénesis de la ATTR y de las limitaciones del TxH ha estimulado el desarrollo de diversos fármacos.
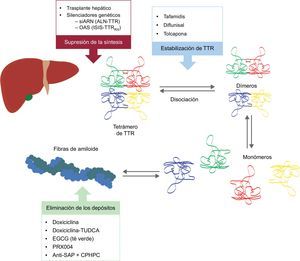
Estos nuevos compuestos actúan en distintos puntos de la cascada de la amiloidogénesis por TTR (figura 6). El tratamiento siempre requerirá la reducción de la proteína precursora, aunque evitar el depósito y eliminar el ya existente serán igualmente importantes. Por ello, creemos que el futuro se dirige hacia un tratamiento combinado.
![Terapias específicas en amiloidosis cardiaca por transtiretina y sus principales dianas. AntiSAP + CPHPC: anticuerpo frente al componente sérico P amiloide + ácido (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic; EGCG: epigalocatequina-3 galato; OAS: oligonucleótidos antisentido; siARN: ARN de interferencia; TTR: transtiretina; TUDCA: ácido tauroursodesoxicólico.](https://static.elsevier.es/multimedia/03008932/0000007000000011/v1_201710271222/S0300893217303500/v1_201710271222/es/main.assets/gr6.jpeg?xkr=eyJpdiI6IjVxNG5PR1lvT3E4M29zWTVjYXMxVkE9PSIsInZhbHVlIjoiSzM3TXlHU2FTOXIxbjdvbG40WTNKSmI1Si9oaytEdU1UZzRRaXhhYTd6Q1lWa0FLOXFwMlpUUi83d1k1TU5kQlhYa2FUQndhWUxUMC90ZnhOYUVxUXBIVkxVNWN0M3dXekhPRmtQeDJMdkFCYmw3d2FYY3VmM2QzbWdzUG83Y2ZHS2VML0xhdmpneVRnMGhuZmY1ZUpNMXlxYndaZ3dpNi92NzAwWStiTUdtVnNNOVRQa084czFUSk1PK25qOFpKNE9rVmxlR1ZURFFuR0dKKzdBci8vZ3lqSVdmYWlNTFRZUGR4UXdzZGxtQVpMYTZ0bWFIaDNicUhEdTQzcG9MVFFVVDhaZzA3Y2JFdk9lcy9IbnJYSWNMYTZkY2VocmdxeTdvTldwNjBwTzA9IiwibWFjIjoiYWJjODRiZjc1NjIwMTQ1MmI1NzU4NTc2YTc0NTZiZGJhYjY0OTFlMzExNGZhN2JlMjczNWYzYzQ4ZTQ0NTVhMyIsInRhZyI6IiJ9 "Terapias específicas en amiloidosis cardiaca por transtiretina y sus principales dianas. AntiSAP + CPHPC: anticuerpo frente al componente sérico P amiloide + ácido (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic; EGCG: epigalocatequina-3 galato; OAS: oligonucleótidos antisentido; siARN: ARN de interferencia; TTR: transtiretina; TUDCA: ácido tauroursodesoxicólico.")
Terapias específicas en amiloidosis cardiaca por transtiretina y sus principales dianas. AntiSAP + CPHPC: anticuerpo frente al componente sérico P amiloide + ácido (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic; EGCG: epigalocatequina-3 galato; OAS: oligonucleótidos antisentido; siARN: ARN de interferencia; TTR: transtiretina; TUDCA: ácido tauroursodesoxicólico.
Dos estrategias están en investigación para inhibir la expresión hepática de TTR: el ARN de interferencia (siARN) y los oligonucleótidos antisentido (OAS).
- •
El siARN lo constituyen moléculas de ARN de doble cadena, que silencian secuencias de ARN mensajero al unirse específicamente a ellas, previniendo la formación de proteínas. El patisirán (ALN-TTR02) ha demostrado una reducción de la producción de TTR de un 80%38. En pacientes con ATTRm la reducción de TTR fue del 87%39. Los primeros ensayos han mostrado resultados prometedores, con estabilidad en los parámetros ecocardiográficos, funcionales y analíticos a 12 y 24 meses40. Los resultados del estudio neurológico de fase III en ATTRm se esperan en 2017 con subanálisis para los pacientes con afección cardiaca (tabla 2). Un segundo agente, el revusirán (ALN-TTR01) —de administración subcutánea y que difiere de patisirán en la nanopartícula lipídica que encapsula el siARN— ha sido objeto de un ensayo clínico en fase III en pacientes con ATTRm que presentan cardiopatía. El estudio se suspendió el año pasado por un aumento inesperado de la mortalidad en el grupo de tratamiento (tabla 2).
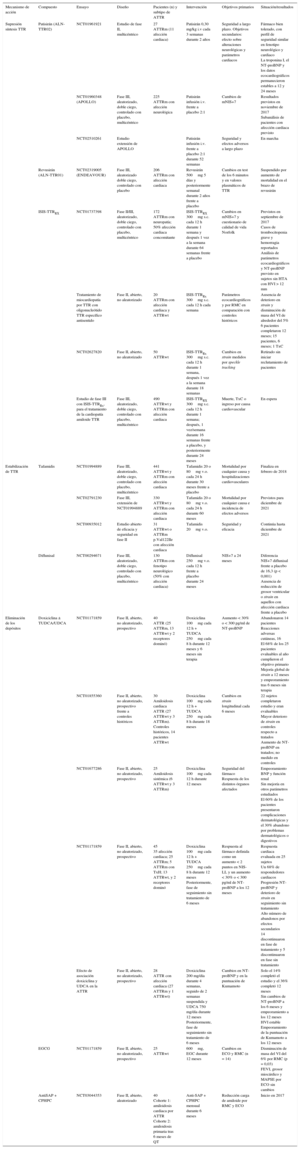
Tabla 2.Principales ensayos clínicos en marcha en amiloidosis cardiaca por transtiretina
Mecanismo de acción Compuesto Ensayo Diseño Pacientes (n) y subtipo de ATTR Intervención Objetivos primarios Situación/resultados Supresión síntesis TTR Patisirán (ALN-TTR02) NCT01961921 Estudio de fase II, multicéntrico 27
ATTRm (11 afección cardiaca)Patisirán 0,30 mg/kg i.v cada 3 semanas durante 2 años Seguridad a largo plazo. Objetivos secundarios: efecto sobre alteraciones neurológicas y parámetros cardiacos Fármaco bien tolerado, con perfil de seguridad similar en fenotipo neurológico y cardiaco
La troponina I, el NT-proBNP y los datos ecocardiográficos permanecieron estables a 12 y 24 mesesNCT01960348 (APOLLO) Fase III, aleatorizado, doble ciego, controlado con placebo, multicéntrico 225
ATTRm con afección neurológicaPatisirán infusión i.v. frente a placebo 2:1 Cambios de mNIS+7 Resultados previstos en noviembre de 2017
Subanálisis de pacientes con afección cardiaca previstoNCT02510261 Estudio extensión de APOLLO Patisirán infusión i.v. frente a placebo 2:1 durante 52 semanas Seguridad y efectos adversos a largo plazo En marcha Revusirán (ALN-TTR01) NCT02319005 (ENDEAVOUR) Fase III, aleatorizado doble ciego, controlado con placebo 206
ATTRm con afección cardiacaRevusirán 500mg 5 días y posteriormente semanal durante 2 años frente a placebo Cambios en test de los 6 minutos y en valores plasmáticos de TTR Suspendido por aumento de mortalidad en el brazo de revusirán ISIS-TTRRX NCT01737398 Fase II/III, aleatorizado, doble ciego, controlado con placebo, multicéntrico 172
ATTRm con neuropatía; 50% afección cardiaca concomitanteISIS-TTRRX 300mg s.c. cada 12 h durante 1 semana y después 1 vez a la semana durante 64 semanas frente a placebo Cambios en mNIS+7 y cuestionario de calidad de vida Norfolk Previstos en septiembre de 2017
Casos de trombocitopenia grave y hemorragia reportados
Análisis de parámetros ecocardiográficos y NT-proBNP previsto en sujetos sin HTA con HVI > 12 mmTratamiento de miocardiopatía por TTR con oligonucleótido TTR específico antisentido Fase II, abierto, no aleatorizado 20
ATTRm con afección cardiaca y ATTRwtISIS-TTRRx 300mg s.c. cada 12 h cada semana Parámetros ecocardiográficos y por RMC en comparación con controles históricos Ausencia de deterioro en strain y disminución de masa del VI de alrededor del 5%
6 pacientes completaron 12 meses; 15 pacientes, 6 meses; 1 TxCNCT02627820 Fase II, abierto, no aleatorizado 50
ATTRwtISIS-TTRRx 300mg s.c. cada 12 h durante 1 semana, después 1 vez a la semana durante 18 semanas Cambios en strain medidos por speckle tracking Retirado sin iniciar reclutamiento de pacientes Estudio de fase III con ISIS-TTRRx, para el tratamiento de la cardiopatía amiloide TTR Fase III, aleatorizado, doble ciego, controlado con placebo, multicéntrico 490
ATTRwt y ATTRm con afección cardiacaISIS-TTRRX 300mg s.c. cada 12 h durante 1 semana; después, 1 vez/semana durante 16 semanas frente a placebo, y posteriormente durante 24 meses Muerte, TxC o ingreso por causa cardiovascular En espera Estabilización de TTR Tafamidis NCT01994889 Fase III, aleatorizado, doble ciego, controlado con placebo, multicéntrico 441
ATTRwt y ATTRm con afección cardiacaTafamidis 20 o 80mg v.o. cada 24 h durante 30 meses frente a placebo Mortalidad por cualquier causa y hospitalizaciones cardiovasculares Finaliza en febrero de 2018 NCT02791230 Fase III, extensión de NCT01994889 330
ATTRwt y ATTRm con afección cardiacaTafamidis 20 o 80mg v.o. cada 24 h durante 60 meses Mortalidad por cualquier causa e incidencia de efectos adversos Previstos para diciembre de 2021 NCT00935012 Estudio abierto de eficacia y seguridad en fase II 31
ATTRwt o ATTRm p.Val122Ile con afección cardiacaTafamidis 20mg v.o. Seguridad y eficacia Continúa hasta diciembre de 2021 Diflunisal NCT00294671 Fase III, aleatorizado, doble ciego, controlado con placebo, multicéntrico 130
ATTRm con fenotipo neurológico (50% con afección cardiaca)Diflunisal 250mg v.o. cada 12 h frente a placebo durante 24 meses NIS+7 a 24 meses Diferencia NIS+7 diflunisal frente a placebo de 16,3 (p < 0,001)
Ausencia de reducción de grosor ventricular o strain en aquellos con afección cardiaca frente a placeboEliminación de los depósitos Doxiciclina ± TUDCA/UDCA NCT01171859 Fase II, abierto, no aleatorizado, prospectivo 40
ATTR (25 ATTRm, 13 ATTRwt y 2 receptores dominó)Doxiciclina 100mg cada 12 h + TUDCA 250mg cada 8 h durante 12 meses y 6 meses sin terapia Aumento < 30% o < 300 pg/ml de NT-proBNP Abandonaron 14 pacientes
Reacciones adversas cutáneas, 16
El 68% de los 25 pacientes evaluables al año cumplieron el objetivo primario
Mejoría global de strain a 12 meses y empeoramiento tras 6 meses sin terapiaNCT01855360 Fase II, abierto, no aleatorizado, prospectivo frente a controles históricos 30
Amiloidosis cardiaca ATTR (27 ATTRwt y 3 ATTRm). Controles históricos, 14 pacientes ATTRwtDoxiciclina 100mg cada 12 h + TUDCA 250mg cada 8 h durante 18 meses Cambios en strain longitudinal cada 6 meses 22 sujetos completaron estudio y eran evaluables
Mayor deterioro de strain en controles respecto a tratados
Aumento de NT-proBNP en tratados; no medido en controlesNCT01677286 Fase II, abierto, no aleatorizado, prospectivo 25
Amiloidosis sistémica (6 ATTRwt y 3 ATTRm)Doxiciclina 100mg cada 12 h durante 12 meses Seguridad del fármaco
Respuesta de los distintos órganos afectadosEmpeoramiento BNP y función renal
Sin mejoría en otros parámetros estudiados
El 60% de los pacientes presentaron complicaciones dermatológicas y el 30% abandono por problemas dermatológicos o digestivosNCT01171859 Fase II, abierto, no aleatorizado, prospectivo 45
35 afección cardiaca; 25 ATTRm; 5 ATTRm con TxH; 13 ATTRwt, y 2 receptores dominóDoxiciclina 100mg cada 12 h + TUDCA 250mg cada 8 h durante 12 meses
Posteriormente, fase de seguimiento sin tratamiento de 6 mesesRespuesta al fármaco definida como un aumento < 2 puntos en NIS-LL y un aumento < 30% o < 300 pg/ml de NT-proBNP a los 12 meses Respuesta cardiaca evaluada en 25 sujetos
Un 68% de respondedores cardiacos
Progresión NT-proBNP y deterioro de strain en seguimiento sin tratamiento
Alto número de abandonos por efectos secundarios
14 discontinuaron en fase de tratamiento y 5 discontinuaron en fase sin tratamientoEfecto de asociación doxiciclina y UDCA en la ATTR Fase II, abierto, no aleatorizado, prospectivo 28
ATTR con afección cardiaca (27 ATTRm y 1 ATTRwt)Doxiciclina 200 mg/día durante 4 semanas, seguido de 2 semanas suspendida y UDCA 750 mg/día durante 12 meses
Posteriormente, fase de seguimiento sin tratamiento de 6 mesesCambios en NT-proBNP y en la puntuación de Kumamoto Solo el 14% completó el estudio y el 36% completó 12 meses
Sin cambios de NT-proBNP a los 6 meses y empeoramiento a los 12 meses
HVI estable
Empeoramiento de la puntuación de Kumamoto a los 12 mesesEGCG NCT01171859 Fase II, abierto, no aleatorizado, prospectivo 25
ATTRwt600mg, EGC durante 12 meses Cambios en ECO y RMC (n = 14) Disminución de masa del VI del 6% por RMC (p = 0,03)
FEVI, grosor miocárdico y MAPSE por ECO sin cambiosAntiSAP + CPHPC NCT03044353 Fase II, abierto, aleatorizado 40
Cohorte 1: amiloidosis cardiaca por ATTR
Cohorte 2: amiloidosis primaria tras 6 meses de QTAnti-SAP + CPHPC mensual durante 6 meses Reducción carga de amiloide por RMC y ECO Inicio en 2017 AntiSAP + CPHPC: anticuerpo frente al componente sérico P amiloide + ácido (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic; ATTRm: amiloidosis hereditaria por transtiretina; ATTRwt: amiloidosis natural; BNP: péptido natriurético cerebral; ECO: ecocardiograma; EGCG: epigalocatequina-3 galato; FEVI: fracción de eyección del ventrículo izquierdo; HTA: hipertensión arterial; HVI: hipertrofia ventricular izquierda; i.v.: intravenoso; MAPSE: excursión sistólica del plano anular mitral; mNIS: Modified Neuropathy Impairment Score; NIS: Neuropathy Impairment Score; NIS-LL: Neuropathy Impairment Score of the Lower Limbs; NT-proBNP: fracción aminoterminal del propéptido natriurético cerebral; TTR: transtiretina; TUDCA: ácido tauroursodesoxicólico; TxC: trasplante cardiaco; TxH: trasplante hepático; QT: quimioterapia; RMC: resonancia magnética cardiaca; s.c.: subcutáneo; UDCA: ácido ursodesoxicólico; VI: ventrículo izquierdo; v.o.: vía oral.
- •
Los OAS son cadenas cortas de oligonucleótidos que se unen específicamente al ARN, previniendo la traslación y la síntesis de la proteína diana4. El ISIS-TTRRx es un OAS de administración subcutánea que demostró una reducción de los valores de TTR, dependiente de la dosis, del 75-90% en voluntarios sanos4. El ensayo de fase III en ATTRm y fenotipo neurológico finalizó en marzo de 2017 y sus resultados se prevén para finales de 2017. Sin embargo, el inicio de un ensayo de fase III en pacientes con ATTRwt y ATTRm con afección cardiaca fue pospuesto por la Food and Drug Administration estadounidense a raíz de algunos casos de trombocitopenia grave en el estudio neurológico (tabla 2). Dado que el 50% de los participantes en el estudio neurológico muestra afección cardiaca, se esperan los resultados de este subestudio a nivel cardiaco para determinar si se retoma la fase III en afección cardiaca. Por otro lado, hay datos preliminares de un ensayo abierto de fase II. En este estudio, 22 pacientes con ATTRwt y ATTRm con afección cardiaca reciben una inyección semanal de ISIS-TTRRx. Según los datos comunicados, el perfil de seguridad del fármaco es muy favorable y los datos intermedios de progresión de la enfermedad a nivel cardiaco por RMC, NT-proBNP y test de 6 minutos son positivos41.
La disociación del tetrámero de TTR en subunidades es un paso crucial en la formación de fibras de ATTR. Diflunisal y tafamidis (2 estabilizadores de TTR) han demostrado hasta ahora su eficacia en la polineuropatía ATTRm.
- •
Tafamidis es una molécula oral que se une a TTR en los sitios de unión de T4 estabilizando la proteína y evitando su disociación. Tras los resultados de un ensayo aleatorizado y doble ciego en 125 pacientes con ATTRm Val30Met en estadios iniciales de afección neurológica42, la Agencia Europea del Medicamento aprobó su uso en 2011 como medicamento huérfano para retrasar la progresión neurológica. Datos recientes demuestran la efectividad del fármaco para lograr la estabilidad neurológica en al menos el 60% de los sujetos tras más de 4 años de seguimiento. Hasta el momento, su uso en la ATTR y la afección cardiológica es limitado. Un estudio de fase II en 21 pacientes con ATTRm y distintas mutaciones demostró estabilidad en NT-proBNP y parámetros ecocardiográficos a los 12 meses43. Datos de otra cohorte a 5 años confirmaron que el fármaco era bien tolerado en la dosis de 20mg, aunque solo unos pocos pacientes con ATTRwt seguían estables a los 3 años y medio44. El estudio ATTR-ACT es un ensayo de fase III cuyo objetivo es evaluar la eficacia, seguridad y tolerancia de las dosis de 20 y 80mg de tafamidis en comparación con placebo en 440 pacientes con ATTRm y ATTRwt e IC durante 30 meses. El objetivo primario incluye mortalidad e ingresos hospitalarios. Sus resultados se esperan en 20183,27.
- •
Diflunisal es un antiinflamatorio no esteroideo que ha demostrado estabilizar la molécula de TTR in vitro. No está disponible en España, pero se puede solicitar medicamente al extranjero para uso compasivo. En un estudio de fase III en ATTRm, con afección predominantemente neurológica y cardiopatía en más de la mitad de los pacientes, los parámetros ecocardiográficos no mostraron diferencias significativas en la evolución (tabla 2)45. Sus potenciales efectos secundarios a nivel gastrointestinal, insuficiencia renal, retención hídrica e hipertensión hacen que su perfil parezca poco adecuado para pacientes con cardiopatía. La evidencia de diflunisal en cardiopatía ATTR es muy limitada y solo cuenta con un único estudio que presenta múltiples limitaciones (no aleatorizado, unicéntrico y con poco seguimiento: 13 pacientes) y en el que no se produjeron ingresos por descompensación de la IC, pero sí un empeoramiento significativo de la función renal46.
- •
Más recientemente, un grupo español ha demostrado como la tolcapona (un inhibidor oral de la catecol-O-metiltransferasa empleado en el tratamiento de la enfermedad de Parkinson) tiene capacidad para unirse in vitro a la TTR de pacientes con ATTRwt y Val122Ile de manera más afín que la descrita con otros estabilizadores47.
Los depósitos de amiloide son muy estables y parecía que el organismo apenas era capaz de eliminarlos. Sin embargo, en los tratamientos que previenen la producción de nuevo amiloide —como la quimioterapia en la AL— se ha observado que los depósitos pueden eliminarse de forma progresiva y a diferentes velocidades según los órganos. Concretamente, a nivel cardiaco, el aclaramiento es especialmente bajo y la evidencia de regresión hasta ahora es escasa. Actualmente, varias moléculas se encuentran en investigación para acelerar el aclaramiento cardiaco de amiloide en la ATTR:
- •
La doxiciclina (antibiótico de uso habitual) interrumpe la formación de fibras de amiloide. De forma sinérgica con el ácido biliar tauroursodesoxicólico (TUDCA) —empleado en el tratamiento de enfermedades hepáticas— demostró efecto en eliminar los depósitos de TTR en modelos animales. Un estudio en fase II con 20 pacientes demostró ausencia de progresión cardiaca o neurológica tras 1 año de tratamiento con doxiciclina/TUDCA, con un perfil de seguridad y tolerancia aceptables4. Otros estudios en fase II han buscado confirmar estos datos empleando la combinación doxiciclina/TUDCA, doxiciclina/ácido ursodesoxicólico o bien doxiciclina aislada48–50. Los resultados preliminares de uno de ellos apuntan a un efecto protector, con un menor empeoramiento de la función cardiaca por strain en el grupo de tratamiento. En la misma dirección se encuentran los hallazgos de otro estudio con 40 pacientes con ATTR en el que se describe una estabilización a 12 meses de parámetros como el NT-proBNP, la clase funcional, la FEVI y el grosor miocárdico (tabla 2). A pesar de estos resultados, todos los trabajos presentaron una alta tasa de abandono (35-44%) sobre todo por efectos secundarios, fundamentalmente fotosensibilidad y alteraciones gastrointestinales (hasta el 30%)48–50.
- •
La EGCG (epigalocatequina-3 galato) —el polifenol más abundante del té verde— ha demostrado in vitro, y en un modelo murino, inhibir los agregados amiloides y la eliminación de depósitos ya formados4. La administración de dosis diarias de 600mg demostró por RMC la estabilización de la masa ventricular izquierda en un grupo de 25 pacientes con ATTRwt (tabla 2)51.
- •
El PRX004 es un anticuerpo monoclonal que actúa uniéndose a epítopos específicos de los monómeros y formas mal plegadas de TTR. De esta forma, promueve la eliminación de los depósitos activando su fagocitosis52. La base de su mecanismo de acción es semejante a la de un anticuerpo empleado en la AL, cuyos estudios en fase II están mostrando resultados favorables. En 2017 está previsto el inicio de un ensayo en fase I-II con este nuevo anticuerpo.
- •
Independientemente de la proteína precursora de amiloide, todos los depósitos contienen el componente P de amiloide sérico (SAP). Utilizando esta molécula como diana, los anticuerpos anti-SAP demostraron promover una reacción mediada por macrófagos y dependiente de complemento que eliminaba de forma masiva depósitos viscerales de amiloide en un modelo murino. El SAP puede ser neutralizado del plasma por el compuesto bis-D-prolina CPHPC y la administración conjunta con IgG anti-SAP permite al anticuerpo alcanzar SAP a nivel de los depósitos tisulares53. Un estudio de fase I publicado en 2015 demostró eliminación de depósitos hepáticos en 15 pacientes con amiloidosis sistémica, sin afección cardiaca, con escasos efectos secundarios53. El inicio de un estudio en fase II con pacientes con amiloidosis cardiaca ATTR y AL está previsto para 2017 (tabla 2).
La amiloidosis cardiaca por ATTR es una entidad que cada vez se diagnostica más frecuentemente. El empleo de técnicas como la gammagrafía con 99mTc-DPD y la RMC permite identificar de forma sencilla y precoz a pacientes con ATTR.
Actualmente varios fármacos se encuentran en las últimas fases de experimentación para el tratamiento específico de esta entidad. Por tanto, creemos que la amiloidosis cardiaca ATTR pasará pronto de considerarse una enfermedad mortal a ser una entidad tratable.
FINANCIACIÓNEste trabajo se ha realizado en parte gracias a ayudas del Instituto de Salud Carlos III y de la Sociedad Española de Cardiología (beca de investigación 2016 a E. González-López). Las ayudas del Instituto de Salud Carlos III cuentan con financiación del Fondo Europeo de Desarrollo Regional «Otra forma de hacer Europa».
CONFLICTO DE INTERESESE. González-López ha participado como ponente en actividades organizadas por Pfizer. P. Garcia-Pavia ha recibido pagos como ponente en actividades organizadas por Pfizer y como consultor de Alnylam, Prothena y Pfizer. E. González-López, A. López-Sainz y P. Garcia-Pavia declaran que Pfizer ha financiado proyectos de investigación de su institución.