
Las mitocondrias son organelos dinámicos, capaces de intercambiar su morfología entre redes elongadas e interconectadas y arreglos fragmentados y desconectados mediante los procesos de fusión y fisión mitocondrial, respectivamente. Estos eventos permiten la transmisión de moléculas de señalización y el intercambio de metabolitos dentro de la célula y participan en una amplia variedad de procesos biológicos, que incluyen el desarrollo embrionario, el metabolismo, la apoptosis y la autofagia. Aunque la mayoría de estos estudios se han realizado en células no cardiacas, la evidencia emergente indica que los cambios en la morfología mitocondrial participan en el desarrollo cardiaco, la respuesta al daño por isquemia-reperfusión, la insuficiencia cardiaca y la diabetes mellitus. En este artículo se revisa cómo la dinámica mitocondrial se altera en diversas enfermedades cardiacas, con especial énfasis en la insuficiencia cardiaca, y cómo este conocimiento podría proporcionar nuevos blancos terapéuticos para su tratamiento.
Palabras clave
La insuficiencia cardiaca es una afección compleja caracterizada por la incapacidad del corazón para responder con un gasto cardiaco acorde a la demanda metabólica del resto de los tejidos. Esta enfermedad representa el estado terminal de las cardiopatías isquémica, valvular o hipertensiva1. En los países industrializados, donde la esperanza de vida de la población y la sobrevida a otras enfermedades cardiacas es elevada, la insuficiencia cardiaca es una enfermedad de alta prevalencia2, 3.
La comprensión de la fisiopatología de la insuficiencia cardiaca ha permitido el establecimiento de la terapia estándar basada en el uso de bloqueadores beta combinado con el bloqueo del sistema renina-angiotensina-aldosterona a través de inhibidores de la enzima de conversión de la angiotensina, antagonistas del receptor de la angiotensina II, antagonistas del receptor de aldosterona y, en algunos casos, la terapia de resincronización4. A pesar de estos avances, la mortalidad anual asociada a insuficiencia cardiaca se mantiene cercana al 10%, y se desconoce la razón por la cual esta enfermedad progresa incluso con una terapia óptima5. Estos antecedentes ponen en evidencia la necesidad de desarrollar nuevos blancos terapéuticos dirigidos a otros mecanismos moleculares y celulares alterados en esta enfermedad.
Durante la década de los cincuenta se estableció, mediante estudios de microscopía electrónica, que las mitocondrias son organelos individuales de doble membrana, cuya membrana interna presenta pliegues característicos denominados crestas mitocondriales6. Esta representación se ha replanteado con base en las evidencias recientes, que muestran que estos organelos conforman una red altamente interconectada y dinámica, cuya morfología varía dependiendo del tipo celular7. Como se muestra en la Figura 1A, las mitocondrias en el corazón adulto se organizan en estructuras discretas y empaquetadas que se disponen a lo largo de las miofibrillas. Esta distribución característica es un fenotipo adquirido durante el desarrollo, dado que la red mitocondrial del cardiomiocito neonato (Figura 1B) se extiende por todo el citoplasma, con una distribución distinta que en el adulto y más parecida a la morfología observada en otras células no cardiacas8.

Figura 1. Morfología mitocondrial del cardiomiocito. A: microscopía electrónica de cardiomiocito de rata neonata en cultivo (panel izquierdo) y tejido cardiaco de rata adulta (panel derecho); en el cardiomiocito neonato, las mitocondrias (M) se encuentran distribuidas en el citoplasma y alrededor del núcleo (N), mientras que en el corazón adulto las mitocondrias se encuentran alineadas entre las unidades sarcoméricas (S). B: microscopía confocal de cardiomiocitos neonatos vivos tratados con la sonda mitocondrial MitoTracker Green; la fotografía central muestra la morfología mitocondrial en condición control; la transducción del cardiomiocito con el adenovirus antisentido para la proteína mitofusina 2 (panel izquierdo) produce fragmentación de la red mitocondrial por una disminución en los procesos de fusión, mientras que la expresión adenoviral de una forma dominante negativa de la proteína relacionada con la dinamina-1 (panel derecho) produce fusión por disminución de los procesos de fisión.
Recientes hallazgos indican que cambios en la morfología mitocondrial podrían ser relevantes en la fisiopatología cardiovascular, pues influyen en la capacidad metabólica celular9. En este artículo se revisa el concepto actual de «dinámica mitocondrial» y cómo se altera en diversas enfermedades cardiovasculares, con especial énfasis en la insuficiencia cardiaca, y se destacan nuevos potenciales blancos terapéuticos.
METABOLISMO CARDIACOPara mantener la función contráctil, el corazón humano adulto normal requiere un suministro diario de trifosfato de adenosina (ATP) equivalente a ∼30kg, alrededor de 70 veces el peso del órgano10. Dado que el contenido de ATP cardiaco es muy bajo (5 μmol/g) y su tasa de hidrólisis es muy elevada (∼30μmol·g−1·min−1 en reposo), todo el ATP se recambia en aproximadamente 10 s, y se sintetiza en un 95% a través de la fosforilación oxidativa11. En el corazón humano adulto, ∼70% del ATP celular deriva de la betaoxidación de ácidos grasos libres (AGL)11.
Los metabolitos generados en la betaoxidación y la glucólisis son incorporados al ciclo de Krebs, y se genera nicotinamida adenina dinucleótido reducido y flavina adenina dinucleótido reducido. Estos equivalentes reductores son oxidados por la cadena transportadora de electrones en la mitocondria, y se genera ATP. Una vez que la energía química se encuentra «almacenada» en el ATP, se transfiere a la creatina por fosforilación mediante la reacción catalizada por la creatincinasa mitocondrial. La fosfocreatina es una molécula de menor tamaño que el ATP, lo que facilita su difusión a través del aparato contráctil de la célula, donde cede su fosfato al difosfato de adenosina (ADP) y regenera ATP en el sitio mismo de utilización. Finalmente, la creatina vuelve a la mitocondria para así iniciar un nuevo ciclo12. En condiciones de alta demanda energética, el ciclo de la creatina permite generar ATP a una tasa 10 veces mayor que la máxima capacidad de la fosforilación oxidativa13.
Tanto estudios clínicos como modelos animales han mostrado que en el corazón insuficiente existen alteraciones en el manejo de los sustratos metabólicos, la producción de ATP y el ciclo de la creatina14. Los pacientes con insuficiencia cardiaca tienen concentraciones cardiacas de fosfocreatina reducidas, lo que genera un estado de depleción energética que se correlaciona con la progresión clínica de la enfermedad14. Estos hallazgos han motivado estudiar el metabolismo energético cardiaco como un nuevo blanco terapéutico.
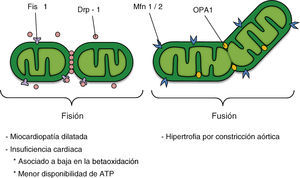
DINÁMICA MITOCONDRIALLas mitocondrias, además de ser el compartimento para numerosas reacciones bioquímicas esenciales en la homeostasis energética, tienen un papel clave en la muerte y el envejecimiento celulares15. Este organelo constituye una red compleja, interconectada y altamente dinámica, mantenida por eventos permanentes, opuestos y balanceados de fusión y fisión mitocondrial9, 16. Tanto el número de túbulos como sus conexiones, así como la distribución subcelular del organelo, son controlados activamente. De esta forma se ha acuñado el término «dinámica mitocondrial» para englobar, al menos, tres procesos distintos: a) el remodelado del retículo mitocondrial mediante procesos de fusión/fisión, el cual se encuentra estrechamente vinculado al estado metabólico celular y es controlado por la actividad de un grupo de proteínas hidrolasas de trifosfato de guanosina (GTPasas) relacionadas con la familia de las dinaminas (Figura 2); b) la motilidad mitocondrial subcelular, particularmente relevante en células polarizadas y que corresponde al desplazamiento de las mitocondrias dependiente de los motores kinesina 1 y 3 y de los adaptadores Milton y Miro17, lo que asegura el suministro local de ATP en procesos biológicos con elevados requerimientos energéticos y el uso de estos organelos como tampones de calcio18, y c) el remodelado de la ultraestructura mitocondrial y la condensación de su matriz, procesos considerados clásicamente como un reflejo del estado metabólico mitocondrial19. Así, un ejemplo de la interrelación entre los distintos estados funcionales de la mitocondria y su ultraestructura es el remodelado de las crestas mitocondriales observado en las transiciones entre los diferentes estados respiratorios o durante la apoptosis20.

Figura 2. Potencial participación de la dinámica mitocondrial en la enfermedad cardiovascular; los factores que regulan la morfología mitocondrial se muestran en la sección superior de la figura. ATP: trifosfato de adenosina; Drp-1: proteína relacionada con la dinamina-1; Fis1: proteína de fisión 1; Mfn 1/2: mitofusinas 1 y 2; OPA1: proteína de la atrofia óptica 1.
LA MAQUINARIA DE FISIÓN MITOCONDRIALLa fisión mitocondrial está regulada en mamíferos por, entre otras, las actividades de la proteína relacionada con la dinamina 1 (Drp-1) y la proteína de fisión 1 (Fis1). Drp-1 posee homología de secuencia con las dinaminas, GTPasas que regulan el tráfico vesicular y la endocitosis21. El mecanismo molecular preciso de las dinaminas y Drp-1 en este proceso es aún materia de debate. Sin embargo, uno de los modelos postula que estas proteínas actúan como mecanoenzimas que participan activamente en el corte de membranas por constricción21.
Drp-1 es una proteína de distribución principalmente citoplasmática, pero con una fracción que se localiza en puntos específicos de la membrana mitocondrial externa que representan futuros sitios de fisión (Figura 2). Drp-1 carece de una secuencia de destino mitocondrial, por lo que se recluta a la membrana a través de Fis1. Esta proteína adaptadora participa en el ensamblaje de complejos de fisión de alta masa molecular21. Actualmente, la interacción directa entre Fis1 y Drp-1 sólo se ha demostrado con proteínas recombinantes, a diferencia de lo observado en levaduras, en las que la localización mitocondrial de Dnm1p (homólogo de Drp-1) se pierde completamente en mutantes para Fis1p (homólogo de Fis1)22. Fis1p recluta a Dnm1p en la mitocondria a través de los adaptadores moleculares Mdv1p y Caf4p23; sin embargo, la existencia de estas proteínas en mamíferos aún no se ha demostrado.
Es importante aclarar que el proceso de fisión mitocondrial ocurre habitualmente en todas las células en condiciones normales. Sin embargo, la fisión mitocondrial también se ha asociado a condiciones de estrés metabólico24, así como a la autofagia25, 26 y la apoptosis27, 28.
LA MAQUINARIA DE FUSIÓN MITOCONDRIALLos principales reguladores de la fusión mitocondrial en el humano son las proteínas mitofusinas (Mfn) y la proteína de la atrofia óptica 1 (OPA1) (Figura 2). Mfn tiene dos isoformas (Mfn1 y Mfn2) que se localizan en la membrana mitocondrial externa con sus dominios N-terminal (dominio GTPasa) y C-terminal orientados hacia el citosol. Mientras que las Mfn 1 y 2 interactúan entre sí para coordinar la fusión de la membrana mitocondrial externa de mitocondrias opuestas29, 30, OPA1 se localiza en el espacio intermembrana y asociada a la membrana mitocondrial interna y participa en el remodelado de las crestas mitocondriales y el acercamiento y la fusión de la membrana mitocondrial interna31, 32. La fusión de ambas membranas mitocondriales parece funcionar como dos eventos independientes y separados. Mientras que la fusión de la membrana externa mitocondrial requiere baja concentración de GTP, la fusión de la membrana interna requiere de la hidrolisis de GTP, además de depender de un potencial de membrana mitocondrial (Ψmt) intacto y, por lo tanto, de una alta síntesis de ATP33.
Evidencias recientes muestran que la fusión regula directamente el metabolismo mitocondrial34. De esta forma, la disminución de la concentración de la proteína OPA1 o de cualquiera de las dos Mfn mediante ARN de interferencia conduce a la formación de mitocondrias fragmentadas con menor consumo de oxígeno y menor Ψmt34. Aunque se conoce la función de estas proteínas (Mfn y OPA1) en la fusión y el remodelado mitocondrial, su relación con la maquinaria metabólica se desconoce, así como por qué la pérdida de algunas de estas proteínas interfiere directamente con la respiración celular. Por el contrario, la sobrexpresión de Mfn2 incrementa directamente la actividad de los complejos respiratorios, la oxidación mitocondrial y la utilización de glucosa35. La expresión de Mfn2 está disminuida en el músculo esquelético de ratas Zucker obesas y en pacientes obesos y diabéticos, lo que destaca la importancia patológica de las alteraciones en la dinámica y la morfología mitocondrial y evidencia que la plasticidad mitocondrial es crítica en el mantenimiento de la función de este organelo36, 37.
DINÁMICA MITOCONDRIAL EN CORAZÓNTejidos que presentan una alta demanda energética, como el corazón y el músculo esquelético, tienden a presentar una red mitocondrial fusionada, mientras que aquellos con baja demanda energética, como el hígado, presentan una red más fisionada38. La dinámica mitocondrial en tejido cardiaco se ha estudiado poco, posiblemente debido a la percepción de que no tiene un papel importante, dada la alta estructuración de la célula cardiaca39.
El cardiomiocito adulto posee 2 grupos de mitocondrias dependiendo de su ubicación citoplasmática. Las mitocondrias interfibrilares (MIF) están intercaladas en la maquinaria contráctil (fibra muscular), mientras que las mitocondrias subsarcolémicas se localizan bajo la membrana plasmática (sarcolema)40. La existencia de estas poblaciones implica que las mitocondrias del tejido cardiaco adulto no forman redes homogéneas como en el cardiomiocito neonato. Curiosamente, en el corazón adulto hay más cantidad de las proteínas de la dinámica mitocondrial que en otros tejidos41.
Debido a la elevada tasa oxidativa de los cardiomiocitos se ha estudiado la posible relación entre la dinámica mitocondrial, el metabolismo y la eficiencia mecánica del corazón. Los estudios de las mitocondrias en el cardiomiocito adulto se han centrado principalmente en evaluar el tamaño y la morfología del organelo en condiciones fisiopatológicas42, 43. Por ejemplo, el daño producido por isquemia-reperfusión se asocia con una morfología mitocondrial fisionada y un aumento en la probabilidad de apertura del poro de transición mitocondrial43. Estos efectos son contrarrestados al inhibir la maquinaria de la fisión mitocondrial, lo cual disminuye el área infartada en ratones sometidos a oclusión de la arteria coronaria43.
De manera similar, en la miocardiopatía diabética existen alteraciones funcionales en la mitocondria, como disminución en la respiración y menor expresión de las proteínas involucradas en la fosforilación oxidativa y producción de ATP42.
En otro estudio de miocardiopatía diabética, se observó que las alteraciones mitocondriales ocurrían mayoritariamente en la subpoblación MIF44. En este modelo experimental, el tamaño, la actividad de los complejos mitocondriales I y III y la concentración de cardiolipina se encontraban reducidos, mientras que la producción de superóxido y la peroxidación lipídica estaban aumentados exclusivamente en las MIF44. Debido a que las MIF generan ATP para sustentar la contracción, la disminución de la actividad metabólica de esta subpoblación podría ser particularmente deletérea contribuyendo a la pérdida de eficiencia mecánica en corazones con miocardiopatía diabética44.
INSUFICIENCIA CARDIACA Y MITOCONDRIADurante el desarrollo de la insuficiencia cardiaca, el corazón experimenta un profundo remodelado metabólico y se modifica su preferencia de sustratos energéticos. Así, a medida que progresa la enfermedad, la oxidación de AGL disminuye paulatinamente45, lo que se asocia a una pérdida progresiva de la cantidad de ATP en el corazón2, 46. En las primeras etapas de la insuficiencia cardiaca aumentan las concentraciones de ADP y AMP, lo que promueve una mayor captación y utilización de la glucosa a través de la activación de la proteincinasa activada por AMP47. Este cambio reduce temporalmente las demandas metabólicas del cardiomiocito; sin embargo, la cantidad de ATP generado a través de la glucólisis es menor que la producida por la betaoxidación, por lo que el contenido de ATP cardiaco inevitablemente disminuye en las etapas terminales de la enfermedad47. Paralelamente, la acumulación de AGL no metabolizados se asocia a un deterioro de hasta un 30% en la eficiencia mecánica del miocardio48.
Las alternativas terapéuticas actuales para la insuficiencia cardiaca buscan potenciar la utilización de glucosa en el corazón inhibiendo directamente la oxidación de AGL o su importe mitocondrial vía carnitina palmitoil transferasa-1. Estas alternativas farmacológicas permitirían compensar la pérdida de ATP en el corazón46, 47. Aunque la mayoría de los fármacos disponibles tienen mecanismos de acción complejos y aún no completamente dilucidados (Tabla 1), todos bloquean la oxidación de AGL y promueven la utilización de glucosa46, 47. Si bien el efecto de estas intervenciones en la insuficiencia cardiaca depende de la etiología o de la etapa de la enfermedad47, varios estudios clínicos indican que la inhibición parcial de la betaoxidación es una alternativa promisoria coadyuvante al bloqueo neurohumoral57, 58.
Tabla 1. Fármacos que modifican la utilización de sustrato en corazón
| Fármaco | Mecanismo propuesto | Efecto | Referencia |
| Etomoxir | Inhibición CPT-1 | Mejora FE y calidad de vida en IC. Requiere más estudios en humanos | Palaniswamy et al 49 ; Schimdt et al 50 |
| Oxfenicina | Inhibición CPT-1 | Inhibe remodelado en modelos animales de IC. Produce hipertrofia dependiente de la dosis por mecanismo desconocido | Greaves et al 51 ; Lionetti et al 52 |
| Perhexilina | Inhibición CPT-1 y CPT-2 | Mejora FE y calidad de vida en IC. Neurohepatotóxico, requiere estudios de administración prolongada | Palaniswamy et al 49 ; Lee et al 53 |
| Ranolazina | Inhibición parcial de betaoxidación | Mejora FE en animales | Palaniswamy et al 49 ; McCormack et al 54 |
| Trimetazidina | Inhibición 3-CAT (CPT-1?) | Mejora FE y calidad de vida en IC. Requiere más estudios en humanos | Palaniswamy et al 49 ; Rosano et al 55 ; Wenmeng et al 56 |
3-CAT: 3-cetoacil coenzima A tiolasa mitocondrial; CPT-1: carnitina palmitoiltransferasa 1; CPT-2: carnitina palmiotoiltransferasa 2; FE: fracción de eyección; IC: insuficiencia cardiaca.
Debido al elevado consumo energético del cardiomiocito, existe una estrecha relación entre la función cardiaca y la mitocondrial. La evidencia clínica señala que el metabolismo energético mitocondrial desempeña un papel crítico en diversas enfermedades cardiacas (Tabla 2).
Tabla 2. Enfermedades cardiacas por trastornos mitocondriales
| Deficiencia primaria | Locus afectado | Fenotipo cardiaco |
| Fosforilación oxidativa | ||
| Mutaciones puntuales en ADNmt | Varios loci | MCD/MCH |
| Pérdida de ADNmt | Varios loci | MCD/MCH |
| Ensamblaje de enzimas COX | SCO2 | MCH |
| SKS/pérdida de ADNmt | Mutación esporádica | MCD |
| Oxidación de ácidos grasos | ||
| Deficiencia en CPT-2 | CPT-2 | MC/Arritmia |
| Deficiencia en SCAD | SCAD | MC infantil |
| Deficiencia en MTP (incluye defectos en LCHAD) | Subunidades MTPa, MTPb | MC infantil/arritmia |
| Deficiencia en VLCAD | VLCAD | MC infantil |
| Deficiencia en CPT-1 | L-CPT-1 | MC |
| Transporte de carnitina | OCTN2 | MC infantil |
| Deficiencia en carnitina traslocasa | CAC | MC infantil/Arritmia |
| Otros | ||
| Ataxia de Friedreich | Frataxina | MCH |
| Síndrome de Barth | G4.5 | MCD |
| Mutación de transportador de fosfato mitocondrial (Mayr et al 60 ) | SLC25A3 | MCH |
| Mutación cadena liviana de la miosina (Poetter et al 61 ) | MYL3 | MCH |
| Mutaciones ARNt mitocondrial | Leu | MCD/MCH |
| Ile | MCH/MC infantil | |
| Lis | MCH |
ADNmt: ADN mitocondrial; ARNt: ARN de transferencia; COX: citocromo c oxidasa; CPT: carnitina palmitoiltransferasa; Ile: isoleucina; LCHAD: 3-hidroxiacil-CoA de cadena larga deshidrogenasa; Leu: leucina; Lis: lisina; MC: miocardiopatía; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MTP: proteína mitocondrial trifuncional; SCAD: deshidrogenasa de acil coenzima A de cadena corta; SKS: síndrome Kearns-Sayre; VLCAD: deshidrogenasa de acil coenzima A de cadena muy larga.
Adaptada de Marín-García et al 59 .
Mutaciones específicas de genes codificantes para proteínas mitocondriales como Ant (translocador de nucleótidos de adenina que intercambia ATP mitocondrial por ADP citosólico), subunidades de los complejos respiratorios, enzimas de la betaoxidación y moléculas relacionadas con el ensamblaje del complejo IV, entre otras, se detectan en pacientes con formas familiares de miocardiopatía dilatada59.
Resultados similares se han replicado en diversos modelos transgénicos, lo que reafirma la relación entre la función mitocondrial y la cardiaca62. La deleción del gen murino que codifica para Ant produce defectos en la fosforilación oxidativa y un fenotipo caracterizado por hipertrofia cardiaca progresiva62. Además, la deleción del factor transcripcional mitocondrial Tfam, esencial para la biogénesis y función del organelo, desencadena una miocardiopatía embrionaria letal, mientras que en los ratones heterocigotos se desarrolla miocardiopatía dilatada severa, y son viables sólo hasta los 20 días de desarrollo63.
INSUFICIENCIA CARDIACA Y DINÁMICA MITOCONDRIALDurante los últimos años han surgido evidencias que vinculan los cambios en la morfología mitocondrial con el desarrollo de enfermedades cardiacas. Tanto en miocardiopatía dilatada64 como en la hibernación miocárdica65, se observan mitocondrias pequeñas y desorganizadas. Hallazgos similares se describen en modelos experimentales de isquemia miocárdica, en los que la red mitocondrial se fragmenta con disminución en el área del organelo junto con un incremento en el número de mitocondrias66.
En diversas miocardiopatías se han documentado cambios en la morfología mitocondrial de los cardiomiocitos. La hipertrofia cardiaca producida por constricción aórtica en la rata se acompaña de aumentos del tamaño mitocondrial y replicación del ADN mitocondrial67. De manera similar, en modelos caninos de insuficiencia cardiaca crónica, el número de mitocondrias pequeñas aumenta, lo que se acompaña de pérdida en la densidad de la matriz mitocondrial68. Esta morfología es similar a la descrita en biopsias musculares de pacientes con trastornos mitocondriales y en modelos de miocardiopatía dilatada, que se caracterizan por la presencia de mitocondrias fragmentadas y una reducción de OPA169. Estos resultados ilustran una asociación entre las alteraciones morfológicas mitocondriales y los cambios en la capacidad bioenergética celular.
Por otro lado, en biopsias de pacientes con miocardiopatía dilatada idiopática se observa la presencia de mitocondrias gigantes con una menor densidad de la matriz mitocondrial, asociado a un aumento en su número70. Resultados similares se han observado en modelos de hipoxia experimental71, lo que indica que la red mitocondrial en el cardiomiocito adulto efectivamente es dinámica y que su remodelado participa en el desarrollo patológico.
Aunque hasta la fecha no se dispone de un modelo integrado que dé cuenta de la participación de la red mitocondrial en la fisiología cardiaca, se ha empezado a estudiar la vinculación entre la dinámica mitocondrial y la enfermedad cardiovascular en los procesos de apoptosis, autofagia, regulación metabólica e isquemia-reperfusión43, que se describen a continuación.
Morfología mitocondrial y apoptosisLa fragmentación de la red mitocondrial en respuesta a estímulos apoptóticos es un hallazgo observado en un gran número de tipos celulares, por lo que se ha postulado una asociación entre la activación de la maquinaria de la fisión mitocondrial y este proceso de muerte celular27, 72. Nuestro grupo ha investigado esta relación en cultivos primarios de cardiomiocitos de rata neonata, que han mostrado en primer término la existencia de la maquinaria molecular de la fisión mitocondrial en las células cardiacas y su relación con los eventos tempranos de la apoptosis inducida por ceramidas28. La pérdida de la integridad de la membrana mitocondrial produce la liberación de citocromo c, endonucleasa G, factor inductor de apoptosis y Smac que activan la vía apoptótica intrínseca73. Este evento está mediado por un proceso complejo que involucra a Drp-1, Mfn2 y la proteína proapoptótica BAX74. La sobrexpresión de las proteínas antiapoptóticas Bcl2 atenúa la apoptosis inducida por isquemia-reperfusión75 y disminuye la probabilidad de apertura del poro de transición mitocondrial frente a sobrecarga de calcio, mejorando el fenotipo contráctil en ratones miocardiopáticos76. Por otro lado, la disminución de la concentración de proteína de fusión Mfn2 se asocia a un incremento en la fragmentación mitocondrial, lo que promueve la liberación de citocromo c y señala a un papel protector de Mfn2 contra la muerte apoptótica inducida por ceramidas en este modelo28.
Morfología mitocondrial y autofagiaLa autofagia es una respuesta celular a condiciones de privación de nutrientes e hipoxia que permite la remoción de organelos y proteínas para la reutilización de aminoácidos y ácidos grasos en procesos celulares fundamentales77. Además, la autofagia cumple un papel en el «control de calidad» celular permitiendo la eliminación de organelos envejecidos y disfuncionales o proteínas dañadas78. La pérdida del Ψmt parece ser la principal señal para la degradación de unidades mitocondriales individuales (mitofagia)25, 79. A pesar de que los procesos de fisión y fusión mitocondrial parecen ser esenciales para la mitofagia80, aún resta por establecer si la regulación de la dinámica mitocondrial se asocia a cambios en el proceso de autofagia en el cardiomiocito26.
Distintos estudios han mostrado que la inhibición de la autofagia en el cardiomiocito promueve su muerte por apoptosis en respuesta a privación de nutrientes o en modelos de isquemia-reperfusión, lo que puede contribuir al deterioro de la función contráctil característica de la insuficiencia cardiaca81, 82.
Morfología mitocondrial y metabolismoComo ya se ha mencionado, las proteínas de la dinámica mitocondrial regulan la función mitocondrial. La disminución en la expresión de Mfn2 se asocia a una reducción en la oxidación de sustratos35, respiración y potencial mitocondrial, hallazgos similares a los descritos para modelos con silenciamiento génico de OPA134 y Drp-183. Estos hallazgos se han replicado parcialmente en modelos animales y en muestras de miocardio de individuos con insuficiencia cardiaca, y se ha observado una significativa disminución de proteínas reguladoras de la dinámica mitocondrial66. En conjunto, estas evidencias indican que cualquier interrupción en la dinámica mitocondrial impacta negativamente en la función de este organelo43. Sin embargo, el papel patogénico de los cambios en la abundancia de estas proteínas y su posible relación con los cambios metabólicos del corazón insuficiente aún no se han dilucidado.
Morfología mitocondrial en isquemia-reperfusión cardiacaLa evidencia previamente señalada indica que el modelo de isquemia-reperfusión corresponde a una condición en la cual los cambios en la morfología mitocondrial son particularmente relevantes. Ong et al43 describieron que la inhibición de la fisión mitocondrial protege al cardiomiocito contra el daño por isquemia-reperfusión en modelos in vitro e in vivo43. El tratamiento con mdivi-1, un inhibidor farmacológico de Drp-1, aumentó la proporción de cardiomiocitos adultos con mitocondrias elongadas y los protegió de la isquemia-reperfusión simulada inhibiendo la apertura del poro de transición mitocondrial y disminuyendo el área infartada43. Estos hallazgos concuerdan con la reducción en la concentración de proteína de fusión OPA1 descrita en muestras de corazones con miocardiopatía isquémica66, que representa la primera evidencia directa de que la modulación de la morfología mitocondrial podría ser una nueva alternativa terapéutica en la enfermedad cardiaca. Sin embargo, el potencial papel protector de esta intervención en la insuficiencia aún no se ha evaluado.
PERSPECTIVASDurante los últimos años la visión clásica de las mitocondrias como unidades discretas e independientes se ha modificado sustancialmente dando paso a un modelo de red mitocondrial, que se remodela dinámica y activamente por procesos de fusión y fisión en respuesta a numerosos estímulos fisiopatológicos. La «dinámica mitocondrial» constituye una parte integral de la flexibilidad metabólica celular. De hecho, a través de estudios de intervención de la maquinaria de dinámica mitocondrial, se ha evidenciado una fuerte asociación entre la morfología y la función de este organelo. En el sistema cardiovascular, hay consenso en que los procesos de dinámica mitocondrial participan en el desarrollo de la enfermedad cardiovascular. La alteración del equilibrio fusión/fisión es una característica común de numerosas enfermedades cardiacas, cuyo punto de confluencia es el síndrome de la insuficiencia cardiaca. Dada la estrecha relación existente entre la dinámica y la funcionalidad de la mitocondria, un blanco terapéutico emergente y complementario al bloqueo neurohumoral es la modulación del metabolismo cardiaco. Estudios iniciales se han basado en la intervención farmacológica del metabolismo de sustratos con resultados promisorios e independientes de la etiología de la insuficiencia cardiaca. Sin embargo, no se ha aclarado si parte del beneficio de las terapias en la insuficiencia cardiaca, en las que se incluirían intervenciones metabólicas, se asocia a cambios en la red mitocondrial. Por otro lado, el desarrollo de intervenciones específicas dirigidas a modular la dinámica mitocondrial podría ser clínicamente relevante y complementario. La investigación exhaustiva de los mecanismos moleculares de esta condición patológica promete revelar nuevas alternativas terapéuticas y es un desafío fundamental para la investigación cardiológica actual.
FINANCIACIÓNJovan Kuzmicic, Andrea del Campo, Camila López-Crisosto, Pablo E. Morales, Christian Pennanen, Roberto Bravo-Sagua, Ramiro Zepeda, Hugo E. Verdejo y Valentina Parra son becarios CONICYT, Chile. Estas investigaciones han sido financiadas por los proyectos FONDECYT 1080436 (Sergio Lavandero), FONDAP 15010006 (Sergio Lavandero), FONDECYT 1090727 (Pablo F. Castro) y FONDECYT 1110180 (Mario Chiong).
CONFLICTO DE INTERESESNinguno.
Autor para correspondencia: Centro Estudios Moleculares de la Célula, Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, Olivos 1007, 8380492 Santiago, Chile. slavander@uchile.cl