El desarrollo de hipertensión arterial pulmonar (HAP) asociada con cardiopatías congénitas (CC) es un proceso multifactorial aún en investigación1. El remodelado vascular que inducen las sobrecargas de presión y volumen en determinadas CC ofrece una explicación satisfactoria de muchos casos, especialmente en el Eisenmenger postricuspídeo. Sin embargo, en un elevado porcentaje de pacientes incluidos en los otros 3 grupos de HAP-CC sospechamos una predisposición individual todavía desconocida, en la que la CC actuaría como mecanismo necesario para el desarrollo de HAP. Los pacientes con cortocircuitos de pequeño tamaño o incidentales representan el mejor ejemplo de esto, ya que la alteración hemodinámica que asocian no es suficiente para inducir por sí sola cambios estructurales en el árbol vascular pulmonar. Estos defectos tienen además el potencial de actuar como válvula de escape para el ventrículo derecho ante una HAP grave, lo que retrasa la aparición de síntomas como el síncope o los relacionados con la insuficiencia cardiaca. En los pacientes con HAP asociada con cortocircuito sistémico-pulmonar, la principal cuestión es identificar en qué pacientes la corrección de este revertirá el proceso y en quiénes el cierre conducirá a la aparición de HAP o la reclasificación del paciente tras la reparación, con peor pronóstico2. La probabilidad de HAP tras la reparación es mayor cuanto más tardía sea la corrección y mayor la lesión vascular preestablecida. No obstante, este subgrupo también se identifica en casos corregidos precozmente, que probablemente tienen una susceptibilidad individual consecuencia de distintos factores, en los que la genética podría ser fundamental.
A continuación, se presenta una familia con 2 casos de HAP-CC portadores de una misma variante no descrita previamente y que determina un fenotipo peculiar: defectos septales diferentes, hemoptisis e inicio precoz. El probando es un varón diagnosticado a los 19 años de HAP junto con una comunicación interventricular (CIV) muscular de 6mm, clasificada como incidental y con cortocircuito predominante derecha-izquierda, descubierto tras un episodio de hemoptisis masiva. Durante este comienzo, se observaron múltiples colaterales bronquiales desarrolladas y el estudio hemodinámico confirmó presiones pulmonares suprasistémicas. Pese a la introducción de triple tratamiento vasodilatador con prostaciclinas sistémicas a altas dosis, el paciente se mantuvo en situación de alto riesgo y finalmente, 4 años tras el diagnóstico, precisó un trasplante bipulmonar en el que se decidió mantener la CIV abierta. A los 10 años del trasplante mantenía buena situación clínica. El segundo caso es la hermana del caso índice, diagnosticada con 26 años de HAP grave junto con comunicación interauricular tipo ostium secundum de 12mm y cortocircuito predominante izquierda-derecha (figura 1). El diagnóstico también se realizó tras un episodio de hemoptisis, en este caso, menor. La paciente inició un tratamiento oral combinado y 4 años después del diagnóstico permanecía en situación de bajo riesgo.
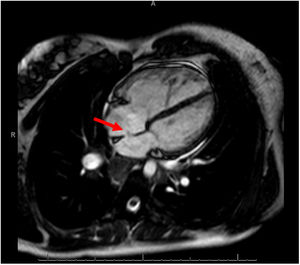
. Destaca la dilatación de la aurícula y el ventrículo derechos.")
Imagen de 4 cámaras en secuencia cine de resonancia magnética cardiaca. Se puede observar un pequeño defecto a nivel del septo interauricular, además de paso de flujo en relación con un cortocircuito sistémico-pulmonar (flecha). Destaca la dilatación de la aurícula y el ventrículo derechos.
Para un mejor estudio etiológico, se ofreció la realización de un panel genes asociados con la HAP. Ambos pacientes, y posteriormente sus familiares, otorgaron su consentimiento por escrito para el estudio, así como su posterior difusión científica. El análisis detectó una variante patogénica en el gen que codifica el receptor de la proteína morfogenética del hueso tipo 2 (BMPR2): NM_001204.6: c.2674delG: p.(Glu892Asnfs*4). No se identificó ninguna otra variante candidata entre el resto de genes incluidos en el panel diseñado por nuestro grupo3. El ulterior estudio de los familiares mostró como esta variante procede de la madre, quien permanecía asintomática, sin datos de hipertensión pulmonar ni defectos septales. Esta mutación no se identificó en el rastreo a los hermanos de la madre y no se pudo explorar en los abuelos, ya fallecidos al momento del estudio por causas diferentes, por lo que se consideró una variante aparentemente de novo (figura 2). Aunque esta variante tampoco figura en las bases de datos de población consultadas (gnomAD genomas, gnomAD exomas, 1000G, ESP, Kaviar, Beacon, Braco), la mayoría de los predictores de patogenicidad in silico aplicados indican un efecto deletéreo de esta, pues genera un codón de parada prematuro.

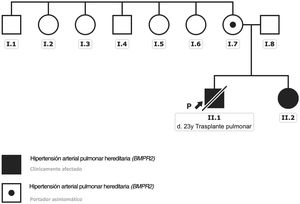
Árbol genealógico de la familia afectada. La flecha negra señala el caso índice. En negro aparecen los pacientes clínicamente afectados y que portan la variante descrita en el gen BMPR2. El punto negro indica a la paciente portadora y clínicamente asintomática. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Nuestra serie describe 2 casos de HAP hereditaria en los que un defecto incidental del tabique podría haber acelerado el desarrollo de vasculopatía pulmonar como factor facilitador. En el área de la susceptibilidad genética para el desarrollo de HAP en pacientes con CC, se ha descrito una prevalencia de variantes patogénicas del gen BMPR2 de hasta el 7,5% en pacientes con HAP relacionada con CC simples (comunicación interauricular, CIV y ductus arterioso persistente) frente al 1,2% de los pacientes con CC sin HAP4. Estudios experimentales han relacionado alteraciones en la vía BMPR2-factor de crecimiento transformador beta (TGFß) con la aparición de defectos septales simples e incluso anomalías septales complejas5. Esta vía metabólica está involucrada en la regulación del crecimiento, la diferenciación y la apoptosis de las células mesenquimales y epiteliales, y se relaciona tanto con la aparición de remodelado vascular adverso como con alteraciones del desarrollo cardiaco. Por otro lado, las mutaciones en BMPR2 son las más frecuentes en la HAP hereditaria, con un patrón de herencia autosómica dominante, penetrancia variable y mayor expresividad en mujeres. Además, estos pacientes suelen presentar una mayor gravedad hemodinámica al diagnóstico y mayor hipertrofia de arterias bronquiales, factores ambos relacionados con la aparición de hemoptisis6. Es interesante constatar que otros genes como TBX4 y SOX17 también se han asociado con el desarrollo de HAP relacionada con CC1.
En definitiva, esta familia pone de manifiesto la relevancia del estudio genético en pacientes con HAP y defectos septales simples, lo cual podía extrapolarse a las distintas formas de HAP-CC. En cuanto a los pacientes con HAP y cortocircuitos incidentales y los pacientes con HAP tras la reparación, el conocimiento genético permite una mejor aproximación al curso de la enfermedad y mejorar su tratamiento y la valoración de familiares. En pacientes con HAP asociada con cortocircuito sistémico-pulmonar amplio, los hallazgos genéticos pueden dirigir la decisión de intervenir el defecto. Los 2 casos presentados son defectos incidentales para los que la decisión de no intervenir quedaba establecida independientemente de la genética. No obstante, conocerlos sí permite predecir la evolución de la enfermedad y diseñar un tratamiento farmacológico particularmente intenso y una correcta valoración familiar.
FINANCIACIÓNEste artículo se financió gracias al proyecto PI 18/01233 «Bases Genético-Moleculares de la Medicina de Precisión en la Hipertensión Arterial Pulmonar», Ministerio de Economía y Competitividad del Gobierno de España (Instituto de Salud Carlos III). A. Cruz Utrilla recibe financiación a través de un contrato Rio Hortega (CM20/00164), Ministerio de Ciencia e Innovación de España (Instituto de Salud Carlos III).
CONTRIBUCIÓN DE LOS AUTORESA. Cruz Utrilla se ha encargado del diseño, escritura y análisis crítico del manuscrito, así como de la inclusión de los pacientes en el estudio. N. Gallego ha colaborado en la realización de estudios genéticos y en la escritura. T. Segura de la Cal ha colaborado en la escritura y análisis crítico. J. Tenorio-Castaño ha colaborado en la realización de estudios genéticos y en el análisis crítico. F. Arribas-Ynsaurriaga ha colaborado en el análisis crítico. P. Escribano Subias ha colaborado en el diseño, escritura y análisis crítico del trabajo.
CONFLICTO DE INTERESESNo existen conflictos de intereses de los autores en relación con este trabajo.