Palabras clave
INTRODUCCION
La monocapa de células endoteliales (CE) que tapiza las paredes vasculares controla la comunicación entre la sangre y los vasos, ejerciendo un papel dual como sensor y transmisor de señales. Las CE son capaces de detectar cambios tanto de tipo físico, relacionados con el estrés mecánico producido por el flujo sanguíneo, la presión arterial o la distensión de la pared, como de tipo químico, debidos a la liberación de sustancias por parte de las células sanguíneas o de los tejidos. Su capacidad de adaptarse funcionalmente a estos estímulos le confiere un papel clave en la regulación de la homeostasis vascular a la cual contribuye mediante la liberación de múltiples sustancias activas (fig. 1). Cuando se produce un desequilibrio en la biodisponibilidad de dichas sustancias que predisponga a la agregación plaquetaria, la trombosis, la inflamación, la vasoconstricción o que produzca un incremento de la permeabilidad vascular, se habla de disfunción endotelial1. En las últimas décadas se ha evidenciado que ciertos factores de riesgo coronario bien caracterizados (LDL, tabaquismo, diabetes, hipertensión...) y otros factores emergentes (radicales libres de oxígeno, homocisteína, infecciones, déficit estrogénico...) producen disfunción endotelial1. Además, de forma experimental se ha demostrado que la función endotelial se ve afectada por numerosos factores vinculados con la cardiopatía isquémica, en especial con los valores de lipoproteínas (LDL, Lp[a]) y con otras proteínas plasmáticas (trombina, plasmina, anticuerpos, etc.).

Fig. 1. Factores derivados del endotelio. Moléculas secretadas por el endotelio en respuesta a estímulos (células circulantes, sustancias vasoactivas, fuerzas físicas) y funciones vasculares reguladas por el endotelio.
El endotelio como barrera de permeabilidad selectiva para macromoléculas
El endotelio de las arterias coronarias y del resto de grandes arterias es de tipo continuo, caracterizado por uniones intercelulares estrechas que restringen el tráfico de macromoléculas. Las CE son células altamente especializadas que se orientan longitudinalmente en la dirección del flujo sanguíneo y que poseen polaridad (superficie luminal en contacto con la luz vascular frente a la superficie estrechamente unida a la membrana basal). El incremento de permeabilidad endotelial parece vinculado a un proceso de contracción celular mediado por calcio y a una desorganización del citosqueleto. Diversos estímulos fisiopatológicos, como la trombina2, generada como consecuencia de la activación de la cascada de la coagulación, o las lipoproteínas3, producen cambios espectaculares en la permeabilidad endotelial. El efecto de la trombina parece ligado a una desorganización del complejo VE-caderina-catenina que forma las uniones intercelulares2.
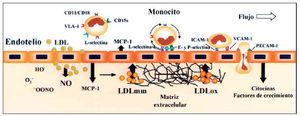
El incremento de la permeabilidad endotelial, producida por las LDL, ha sido observado in vitro4, ex vivo5 e in vivo6. De hecho, hasta hoy la única forma de inducir lesiones ateroscleróticas en animales de experimentación, similares a las encontradas en las arterias humanas, es mediante la administración de dietas ricas en colesterol y grasa saturada. Este tipo de dietas produce un aumento de las concentraciones de LDL en plasma y facilita su acumulación en el espacio subendotelial en zonas donde la permeabilidad se halla incrementada (fig. 2). En estos modelos animales se ha observado que las regiones más propensas a desarrollar lesiones ateroscleróticas presentan una mayor permeabilidad a las LDL y las VLDL7. Este efecto de las lipoproteínas parece vinculado a la desorganización que producen en el citosqueleto celular8,9, en el que se ha involucrado a las proteínas Rho9.

Fig. 2. Disfunción endotelial en la aterogénesis. Las LDL interaccionan con componentes de la matriz extracelular (proteoglucanos y glucosaminoglucanos) presentes en la íntima lo que favorece su degradación proteolítica y su oxidación. Las LDLox per se inducen la expresión de factores quimiotácticos (MCP-1) y de moléculas de adhesión (VCAM-1 y P-selectina), que son clave en el proceso de reclutamiento de monocitos. Los leucocitos ruedan sobre la superficie endotelial y se unen primero débilmente a las selectinas, y posteriormente con más fuerza a CAM de la familia de las inmunoglobulinas (ICAM y VCAM), cuyos valores de expresión se encuentran incrementados en las áreas de lesión.
Finalmente, se ha observado que concentraciones aterogénicas de LDL nativas y bajas concentraciones de LDL oxidadas (LDLox) incrementan la permeabilidad vascular, ya que reducen el contenido de proteoglucanos de heparan sulfato que componen la matriz extracelular del espacio subendotelial. Este efecto se produciría mediante una regulación negativa de la síntesis de estas moléculas, así como de un incremento de su degradación gracias a la inducción de la secreción endotelial de heparanasa10.
El endotelio como regulador de la respuesta vascular a estímulos inflamatorios
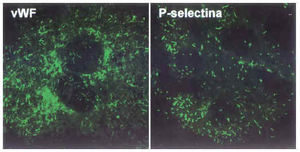
La arteriosclerosis presenta características de enfermedad inflamatoria crónica, y los resultados obtenidos en modelos animales, en los que se induce el desarrollo de lesiones ateroscleróticas, evidencian la relevancia del endotelio en el desarrollo y la perpetuación de dicho estado inflamatorio. El endotelio activado expresa/secreta citocinas (como la interleucina-1 [IL-1]), factores de crecimiento (PDGF, bFGF...), factores quimioatrayentes (proteína-1 quimiotáctica para monocitos [MCP-1]) y proteínas de superficie que actúan como moléculas de adhesión (CAM) de leucocitos circulantes11,12. Cybulski y Gimbrone, en un estudio pionero llevado a cabo en conejos hipercolesterolémicos, demostraron que en el endotelio de las áreas donde se infiltran monocitos y se desarrollan lesiones arterioscleróticas se detectaban elevados valores de una de estas CAM, la molécula de adhesión vascular-1 (VCAM-1), cuya expresión es indetectable en el endotelio normal13. Otra de las CAM de la que se dispone de mayor evidencias experimentales sobre su papel en la adhesión de monocitos es la P-selectina. La P-selectina, que está almacenada en los cuerpos de Weibel-Palade junto con el factor de von Willebrand (vWF) (fig. 3), se expone en la superficie de las CE de las lesiones ateroscleróticas pero no en áreas sin lesión14.

Fig. 3. Moléculas de adhesión en CE en cultivo. Las CE en cultivo crecen en monocapa y adquieren una disposición típica en forma de «adoquinado». Se muestra la tinción de dos proteínas implicadas en la adhesión: el factor de von Willebrand (vWF) y la P-selectina, ambas se almacenan en los cuerpos de Weibel-Palade y son expuestas/secretadas cuando la célula es estimulada.
El dominio extracelular de las CAM (tabla 1) puede liberarse al torrente circulatorio, y parece que sus valores circulantes se correlacionan con la expresión a nivel celular. Por ello, actualmente se evalúan dichos valores como marcadores de evolución de las lesiones ateroscleróticas y procesos patológicos asociados (diabetes, dislipemias, hipertensión y reestenosis postangioplastia). En general, estas enfermedades producen un aumento de las concentraciones de las formas solubles de algunas de las CAM mencionadas. Se han encontrado valores elevados de las formas solubles de ICAM-1 y P-selectina en pacientes con cardiopatía isquémica15,16, y de ICAM-1 y VCAM-1 en pacientes con hipertrigliceridemia17 y enfermedad arteriosclerótica periférica o cerebral18,19. En el Physicians' Health Study los valores circulantes de ICAM-1 en el momento de la selección de los pacientes predijo el desarrollo de episodios cardiovasculares a largo plazo, y su correlación con otros marcadores de inflamación como los valores de proteína C reactiva20. Recientemente, nuestro grupo ha evidenciado que el tratamiento con inhibidores de la HMG-CoA reductasa mejora la función endotelial de pacientes con hipercolesterolemia familiar heterozigota, y reduce significativamente las concentraciones circulantes de E-selectina21 (fig. 4). Por tanto, en estos pacientes la mejora de la respuesta vasodilatadora dependiente del endotelio parece, además, asociada a una disminución de la activación/lesión endotelial.


Fig. 4. Valores de E-selectina circulantes (sE-selectina) en pacientes con hipercolesterolemia familiar en el momento de comenzar el tratamiento con simvastatina (basal) y después de 12 y 52 semanas de ser instaurado el tratamiento. (Modificado de Alonso et al21.)
El endotelio como regulador del tono vascular: papel del óxido nítrico
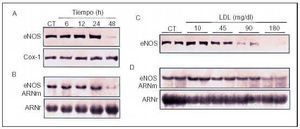
Desde su descubrimiento y caracterización22,23, el óxido nítrico (NO) se ha revelado como la molécula más versátil que sintetiza el endotelio, ya que posee la mayor parte de las propiedades ateroprotectoras que se le atribuyen a éste: vasodilatador, antiagregante plaquetario, inhibidor de la proliferación de las CML, antioxidante e inhibidor de la expresión de CAM y la adhesión de monocitos. Por tanto, a través de la alteración de la producción de NO los estímulos aterogénicos perturban profundamente la homeostasis vascular y potencian el desarrollo de lesiones ateroscleróticas. Esta disminución de la dilatación dependiente del endotelio es la manifestación más temprana de la disfunción endotelial. Se observa tanto en pacientes con hipercolesterolemia como en aquellos con valores elevados de Lp(a), diabetes u homocistinuria24,25. La alteración de la dilatación dependiente del endotelio producida por la hipercolesterolemia se debe a una disminución de la biodisponibilidad de NO26. Las LDL pueden alterar la producción de NO actuando de diferentes formas: incrementando la fracción de la enzima que regula la producción de NO (la óxido nítrico sintasa endotelial, eNOS) unida a caveolina-1, y por tanto insensible a la regulación por calcio-calmodulina27, incrementando la degradación del NO28, o bien aumentando la inhibición competitiva de la formación de NO por ADMA (asymmetric dimethylarginine), un inhibidor endógeno cuyos valores se encuentran elevados en pacientes hipercolesterolémicos29. Sin embargo, in vivo la hipótesis más verosímil parece ser una reducción neta de la actividad de la enzima eNOS debido a una inhibición de las concentraciones de ARNm y la proteína de esta enzima. Esta inhibición se ha observado in vitro en respuesta a LDLox30 y concentraciones aterogénicas de LDL nativas31,32,33 (fig. 5). Además, se ha observado que las arterias humanas con lesiones ateroscleróticas contienen menor cantidad de eNOS34.

Fig. 5. Efecto de concentraciones aterogénicas de LDL sobre los valores de ARN mensajero (ARNm) y proteína de la enzima eNOS. A) Western blot que muestra la inhibición dependiente de tiempo de los valores de la enzima eNOS en CE humanas por concentraciones aterogénicas de LDL (180 mg/dl). En estas condiciones no se vieron afectados los valores de ciclooxigenasa-1 (Cox-1), enzima reguladora de la producción de prostaciclina. B) Northern blot que pone de manifiesto la inhibición dependiente del tiempo de los valores de ARNm de eNOS. C y D) Inhibición dependiente de la dosis de los valores de proteína (C) y ARNm (D) de eNOS por las nLDL. (Modificado de Vidal et al32.)
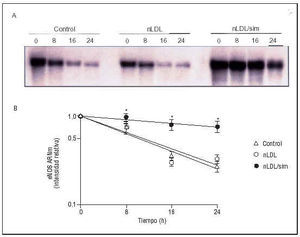
La mejoría de la función endotelial producida por los tratamientos con fármacos hipolipemiantes35,36 ha llevado a un intenso estudio de los mecanismos moleculares que subyacen a tales efectos. Actualmente, varios grupos de investigación han puesto de manifiesto en CE en cultivo que la temprana mejoría de la dilatación dependiente del endotelio producida por los inhibidores de la HMG-CoA reductasa se debe a un incremento de la vida media del ARNm que codifica para la enzima eNOS33 (fig. 6).

Fig. 6. Incremento de la vida media del ARN mensajero de eNOS por inhibidores de la HMG-CoA reductasa. A) CE humanas fueron incubadas con LDL (LDL, 180 mg/dl) en ausencia o en presencia de simvastatina (sim, 0,1 µmol/l) durante 48 h y, posteriormente, durante tiempos variables (de 8 a 24 h) en presencia de un inhibidor de la transcripción. Se ilustra un experimento representativo de Northern blot donde se aprecia cómo la simvastatina incrementa significativamente la vida media del mensajero de eNOS. B) Resultado de la cuantificación de los experimentos de Norther blot. *< 0,05 frente a control. (Modificado de Martínez-González et al33.)
El endotelio como regulador del equilibrio hemostático/trombótico
Durante muchos años la incapacidad del endotelio de activar la cascada de coagulación y fomentar la adhesión de plaquetas se consideró una función pasiva, relacionada con ciertas carencias más que como resultado de su participación activa en la hemostasia. Esta idea cambió al descubrirse que las CE producían prostaciclina (PGI2), un extraordinario inhibidor de la agregación plaquetaria. Posteriormente, se descubrió que el NO actúa sinergísticamente con la prostaciclina como antiagregante plaquetario23. De hecho, el NO inhibe la adhesión, la activación, la secreción y la agregación plaquetaria.
El endotelio ejerce un papel central en la regulación de la hemostasia ya que aporta importantes elementos de los sistemas de coagulación, trombosis y fibrinólisis del organismo. Además de NO y PGI2, las CE producen trombomodulina, moléculas con actividad heparina-like y ADPasa, que hidroliza el ADP (agregante plaquetario). Como agentes protrombóticos secreta PAF, moléculas de adhesión para las plaquetas (como vWF, fibronectina y trombospondina) y factores de coagulación (como el factor V) y, en respuesta a distintos factores fisiopatológicos expresa factor tisular37.
El endotelio también regula la fibrinólisis, ya que produce activador tisular del plasminógeno (t-PA), urocinasa e inhibidor-1 del t-PA (PAI-1). La biosíntesis de estas moléculas es alterada por los lípidos plasmáticos, particularmente por las VLDL38, que ejercen su acción a través de un elemento de respuesta a VLDL presente en el promotor del gen que codifica para el PAI-139. Este efecto de las VLDL se incrementa si éstas proceden de individuos con hipertrigliceridemia39. La susceptibilidad individual a la alteración de esta función endotelial por la hipertrigliceridemia parece ligada a la presencia de ciertas variantes polimórficas en el gen del PA-1, como el polimorfismo HindIII que se ha asociado con diferencias en la actividad y la capacidad de las VLDL de modular la producción de PAI-140. Algunos estudios epidemiológicos han encontrado una asociación entre los valores elevados de colesterol en plasma y tiempo prolongado de euglobina, lo que sugiere una alteración del equilibrio entre la liberación de activadores del plasminógeno y sus inhibidores por el endotelio41. El efecto de los inhibidores de la HMG-CoA reductasa sobre el equilibrio t-PA/PAI-142,43 podría contribuir a los efectos vasculares directos que se han atribuido a estos fármacos.
Finalmente, los resultados de diferentes estudios epidemiológicos en relación con el posible efecto de los lípidos sobre la producción y la secreción del vWF son contradictorios. Unos estudios sugieren que el tamaño de las partículas de LDL es determinante de los valores de vWF circulante41; otros han encontrado una correlación entre los valores circulantes de vWF con las cifras de triglicéridos plasmáticos y HDL, pero no con los de colesterol total44; mientras que el estudio EURODIAB únicamente encontró una correlación de los valores de vWF con el colesterol total y los triglicéridos en varones, pero no en mujeres45.
Significación fisiopatológica del equilibrio proliferación/apoptosis en el endotelio
En condiciones normales las CE tienen un índice de recambio muy bajo, que aumenta significativamente en las zonas más vulnerables a la aparición de lesiones, donde también se observa un mayor número de células en proceso de apoptosis. De hecho, el flujo laminar, que se considera uno de los factores endógenos de mayor poder antiaterogénico, protege a las CE inhibiendo la apoptosis46. Por el contrario, algunos factores proaterogénicos, como las LDLox47, las citocinas inflamatorias, la angiotensina II y las especies reactivas de oxígeno, inducen apoptosis de las CE48. Además, recientemente hemos observado que las LDL nativas a concentraciones aterogénicas promueven per se apoptosis de las CE49. Por tanto, los valores circulantes de LDL modulan profundamente la fisiología del endotelio vascular y pueden condicionar la capacidad de respuesta de éste a estímulos proaterogénicos vinculados con otros factores de riesgo.
La relevancia del endotelio en la homeostasis de la pared vascular se evidencia de forma espectacular en las intervenciones intravasculares que causan desendotelización, con la consiguiente pérdida temporal de las funciones vasoprotectoras que éste realiza. La pérdida del endotelio y la exposición del contenido de las placas ateroscleróticas, sobre todo el componenete lipídico50, activa la adhesión/agregación de plaquetas, que liberan localmente factores quimiotácticos y mitogénicos que ponen en marcha la reparación de la pared vascular51. Aunque los mecanismos moleculares implicados en la recuperación de un endotelio funcional se activan de forma inmediata, la exposición de la pared desprovista de endotelio puede persistir durante varias semanas52. En la reparación vascular se implican las CE, que a partir de los bordes del endotelio intacto colonizan las áreas contiguas desendotelizadas y las CML, que proliferan y secretan matriz extracelular53.
La reendotelización se activa por la pérdida de la inhibición por contacto de la replicación, que mantiene al endotelio inactivo, y por la liberación local de factores que específicamente potencian dicha actividad, como el factor de crecimiento de endotelio vascular (VEGF)54 y el factor de crecimiento derivado de fibroblastos (FGF)55. El primero de ellos es sintetizado por las CML y ejerce un efecto trófico de forma relativamente específica sobre las CE, mientras que el FGF lo producen y almacenan tanto las CE como las CML, y ejerce efectos sobre ambas. El efecto de estos factores, particularmente del FGF, puede prolongarse en el tiempo ya que puede permanecer activo unido a proteínas de matriz extracelular. El NO también desempeña un papel en los mecanismos de reparación vascular. El NO estimula la migración y la proliferación de las CE56, lo que juntamente con su efecto inhibitorio sobre la migración y la proliferación de las CML57, facilita la reendotelización y limita la proliferación neointimal ligada a la lesión vascular. De hecho, en modelos experimentales se ha observado que las áreas que reendotelizan antes presentan un menor grado de engrosamiento intimal y una proliferación de las CML58.
Regulación de la función endotelial mediante el control de la expresión génica
La disfunción endotelial asociada a la aterogénesis implica una alteración profunda de su patrón de expresión génica que conlleva la inducción de genes que en condiciones fisiológicas estarían reprimidos, así como la inhibición de otros expresados en condiciones normales. Uno de los principales moduladores de la expresión endotelial de genes es el régimen de flujo al que está expuesto el endotelio. Las lesiones ateroscleróticas humanas se localizan preferentemente en las bifurcaciones y curvaturas de las arterias, donde el flujo sanguíneo es lento u oscilatorio59. Por el contrario, las regiones de flujo laminar uniforme parecen estar relativamente protegidas del desarrollo de lesiones60. En los últimos años se ha comenzado a comprender los mecanismos que subyacen en la modulación de la función endotelial por fuerzas mecánicas. Utilizando técnicas de análisis diferencial de la expresión génica se han identificado varios genes que son específicamente inducidos por flujo laminar, como la ciclooxigenasa-2 y la eNOS61. Por el contrario, en condiciones de flujo turbulento no se produce la activación de dichos genes. El flujo prácticamente modula todas las moléculas producidas por el endotelio: moléculas que regulan el tono vascular (NO, endotelina-1), el crecimiento celular (FGF, PDGF-AA y -BB, TGF-β), la adhesión (MCP-1, VCAM-1, ICAM-1), la fibrinólisis (t-PA) y la trombosis (trombomodulina y factor tisular)62-64. Los mecanismos responsables de la modulación por flujo están siendo objeto de estudio. Parece que estos efectos se deben, al menos en parte, a la presencia en el promotor de estos genes de elementos de respuesta a flujo (shear stress response elements, SSRE)65. Además, el flujo modula la activación de múltiples factores de transcripción (NF-κβ, Egr-1, c-jun, c-fos) implicados en la activación/represión de los genes mencionados anteriormente66.
En los últimos años también se han acumulado evidencias que subrayan la relevancia el factor nuclear kappa beta (NF-κβ) como común denominador en la expresión coordinada de los genes inducidos en la activación endotelial67. El factor NF-κβ se encuentra en el citoplasma en forma de heterodímero inactivo unido a proteínas inhibidoras denominadas genéricamente I-κβ. Cuando la célula recibe estímulos inflamatorios se activa la fosforilización y ubiquitinación de I-κβ, lo que sirve de «señal» para que sufra degradación proteolítica. Entonces, los heterodímeros se translocan al núcleo donde activa la transcripción de genes diana que poseen en su promotor elementos de respuesta κβ. Entre los genes regulados por NF-κβ se encuentran las citocinas (factor necrosante de tumores [TNF-α e IL-1, -6 y -8]), los factores estimuladores de la formación de colonias de granulocitos/macrófagos (G-CSF, M-CSF, GM-CSF), MCP-1, el factor tisular, varias moléculas de adhesión (ICAM-1, VCAM-1) y c-myc.
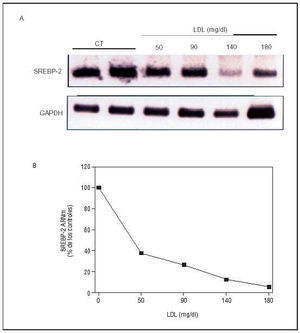
Sin embargo, la disfunción endotelial puede involucrar a otros factores de transcripción. En este sentido, en un reciente estudio realizado en un modelo porcino de hipercolesterolemia, mediante técnicas de análisis diferencial de expresión génica hemos puesto de manifiesto que las LDL, a través de la regulación de factores de transcripción, como las proteínas de unión a elementos de regulación por esteroles (sterol regulatory element binding proteins, SREBP), modulan la expresión de enzimas implicadas en la síntesis endógena de colesterol, tanto en CE en cultivo como en la pared vascular in vivo68 (fig. 7). Dado que el número de genes regulados por SREBP es muy amplio e incluye algunos de notable interés en el desarrollo de lesiones ateroscleróticas, como los receptores de las LDL69 y las HDL70, la lipoproteinlipasa71 y PPARγ72, las LDL nativas podrían afectar a otros muchos genes a través de la modulación de la actividad de estos factores de transcripción.

Fig. 7. Modulación de la expresión endotelial de SREBP-2 por las LDL. A) Inhibición dependiente de la dosis de los niveles de ARNm de SREBP-2 (principal factor de transcripción implicado en la regulación génica mediada por colesterol) por LDL en CE. B) Gráfica que ilustra la cuantificación de los v(Modificado de Rodríguez et al68.)
Correspondencia: Prof. Lina Badimón.
Laboratorio de Investigación Cardiovascular.
Hospital de San Pa blo.
Avda. S. Antonio M. Claret, 167. 08025 Barcelona.
Correo electrónico: ibmucv@cid.csic.es