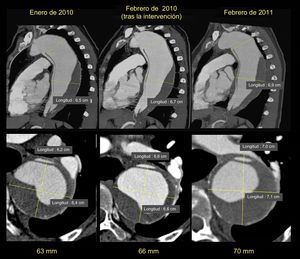
Se presenta el caso de una paciente de 55 años, sin antecedentes de interés, que en agosto de 2008 tuvo una disección aórtica tipo B. La tomografía computarizada mostró una amplia puerta de entrada de la disección después de la salida de la subclavia izquierda extendiéndose hasta la bifurcación iliaca. La aorta torácica descendente estaba dilatada (60 mm) con trombosis parcial de la falsa luz. La válvula aórtica era trivalva y normofuncionante, y la aorta ascendente era de tamaño normal. Ante la ausencia de complicaciones agudas, se decidió tratamiento con bloqueadores beta y se planteó diferir el tratamiento endovascular. Dado que el arco aórtico tenía un ángulo agudo y la distancia entre las arterias subclavia y la carótida izquierda era corta, no existía un cuello suficiente para el implante proximal de la endoprótesis, por lo que se indicó una intervención en dos tiempos: un bypass aortobicarotídeo y después el tratamiento endovascular. En febrero de 2010 se procedió al bypass aortobicarotídeo, complicado con una disección retrógrada de aorta ascendente desde el punto de pinzamiento, por lo que se recambió la aorta ascendente media y distal con una prótesis vascular Hemashield 28. El seguimiento mostró dilatación progresiva de la aorta torácica descendente (figura 1), a pesar de la cual la paciente se negó a intervención alguna.

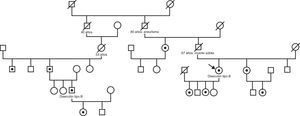
En agosto de 2011, la paciente refirió que el hijo de un primo segundo, de 38 años, había tenido una disección aórtica tipo B. Se trataba de un varón sin antecedentes de interés que había ingresado en otro centro por una disección aórtica con origen tras la salida de la subclavia izquierda y progresión hasta la bifurcación iliaca. En el momento de la disección, había una dilatación ligera de la aorta torácica descendente (39 mm), con una válvula aórtica trivalva y normofuncionante y sin dilatación del resto de los segmentos aórticos. Se reorientó el caso como enfermedad aórtica familiar; se realizó una exploración física minuciosa de ambos pacientes, que no mostró estigmas de síndrome de Marfan ni Loeys-Dietz y se solicitó análisis genético del gen ACTA2 (que codifica la isoforma 2 de la actina alfa en células musculares lisas vasculares) del segundo paciente. El análisis mostró una mutación en heterocigosis (c.253G>A) con cambio de aminoácido (p.Glu85Lys) no descrita previamente. La misma mutación se detectó también en el caso índice y en el padre, el tío paterno y la hija del segundo caso y en la hermana, la tía paterna, las hijas y la sobrina del caso índice (figura 2). Ninguno de los portadores de la mutación en ACTA2 presentaba iris flocculi o livedo reticularis, hallazgos descritos previamente1. Ante la enfermedad aórtica familiar y la fragilidad aórtica evidenciada durante el acto quirúrgico, se decidió evitar tratamiento endovascular y se sometió a la paciente a cirugía abierta para recambio de la aorta torácica descendente por un tubo protésico con reimplantación de los troncos viscerales.

A pesar de que la mayoría de las disecciones aórticas tipo B se presentan en pacientes de mediana edad con hipertensión arterial y/o arteriosclerosis, existe una proporción no despreciable de pacientes con presentación precoz de probable base genética no bien conocida. En ausencia de un síndrome identificable, hasta el 21,5% de los pacientes con aneurismas de aorta tienen antecedentes familiares; se identifican mutaciones genéticas en un 20%2, y la mutación en ACTA2 es la más frecuente (10-14%)3. Esta mutación se asocia a enfermedad aórtica con una penetrancia del 48% y expresividad variable, más frecuentemente en forma de disecciones aórticas tipo A, incluso con diámetros aórticos no avanzados, y casos aislados de disecciones tipo B4; también se ha asociado a enfermedad coronaria y cerebrovascular prematura1.
A pesar de que el cribado familiar con técnicas de imagen de familiares de primer grado de pacientes con enfermedad aórtica prematura puede ser razonable, las indicaciones para el análisis genético no están bien establecidas. La guía de práctica clínica de la American Heart Association/American College of Cardiology recomienda la búsqueda de mutaciones en ACTA2 en caso de agregación familiar de enfermedad aórtica5. No obstante, es importante destacar que la penetrancia es baja y la expresividad variable, lo que dificulta el seguimiento de los portadores asintomáticos de la mutación.
Un aspecto destacable del presente caso es la importancia de la sospecha diagnóstica de enfermedad aórtica familiar en pacientes jóvenes con enfermedad aórtica; esta circunstancia debe hacer insistir en el interrogatorio sobre los antecedentes familiares y descartar casos sindrómicos. Otro punto importante es que el diagnóstico de enfermedad aórtica familiar hace razonable (sin el respaldo de ensayos clínicos) cambiar la estrategia terapéutica. De manera similar que en el síndrome de Marfan y otras conectivopatías6, implantar una endoprótesis vascular puede estar contraindicado si no es como terapia puente a la cirugía. Finalmente, se debe tener presente que la enfermedad aórtica familiar puede presentarse afectando solo a la aorta descendente y que el riesgo de disección está presente incluso sin una marcada dilatación aórtica.
A la Sra. Francesca Huguet, enfermera de la Unidad de Marfan, por su imprescindible colaboración.