
Los pacientes con insuficiencia renal crónica tienen más probabilidad de morir por causa cardiovascular que de progresar a insuficiencia renal terminal. La excreción urinaria de albúmina es un marcador de riesgo tanto de enfermedad renal como de enfermedad cardiovascular en diabéticos y no diabéticos. En la insuficiencia renal crónica, la hipertensión, la dislipemia y la diabetes mellitus son los principales factores de riesgo de disfunción endotelial, inflamación, estrés oxidativo y arteriosclerosis acelerada. La inhibición del sistema renina-angiotensina enlentece la progresión de la enfermedad renal en la diabetes mellitus tipos 1 y 2 y en los no diabéticos con nefropatía. El grado de reducción de albuminuria se relaciona con el riesgo cardiovascular en la diabetes mellitus tipo 2 y la nefropatía. La excreción urinaria de albúmina debe ser utilizada como objetivo terapéutico similar a la presión arterial para reducir las complicaciones renales y cardiovasculares en pacientes con nefropatía.
Palabras clave
La enfermedad cardiovascular (ECV) acelerada es una complicación frecuente de la enfermedad renal (ER). La insuficiencia renal crónica (IRC) se asocia con un significativo incremento de riesgo de morbimortalidad cardiovascular (CV) al margen de la presencia de factores de riesgo cardiovascular (FRCV) tradicionales, como la diabetes mellitus (DM), la hipertensión arterial (HTA), la concentración de lipoproteínas y el hábito tabáquico, hasta el punto de que se la considera FRCV independiente, según la National Kidney Foundation, la American Heart Association y el Seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure1.
Los pacientes con IRC muestran mayor prevalencia de insuficiencia cardiaca congestiva (ICC), cardiopatía isquémica (CI) en cualquiera de sus formas clínicas, incluida la isquemia silente, arritmias ventriculares complejas, fibrilación auricular (FA), hipertrofia ventricular izquierda (HVI), rigidez y calcificación arterial y calcinosis del anillo mitral y de la válvula aórtica. La reducción del filtrado glomerular (FG) predice la incidencia de episodios isquémicos2. porque refleja más probabilidad de muerte por proceso CV que por insuficiencia renal3,4, en parte por no recibir el tratamiento cardioprotector adecuado al riesgo5. La mitad de los pacientes con insuficiencia renal avanzada o terminal (IRT) mueren por causa CV4-7, que es de 15 a 30 veces más elevada que la de la población general tras los ajustes por edad4,8, más en jóvenes (25–34 años), en los que la mortalidad CV es 500 veces mayor que la de los controles3.
Este proceso progresa a través de varios mecanismos como HVI, rigidez arterial, inflamación, disfunción endotelial, estrés oxidativo y formación precoz de placas de ateroma. El 40% de los pacientes que inician tratamiento de hemodiálisis (HD) tienen afección coronaria y el 85%, alteraciones en la estructura y/o la función del ventrículo izquierdo (VI)7. La prevalencia de HVI es del 45,2% de los pacientes con aclaramiento de creatinina (aCr)<25ml/min; del 30,8% de los que tienen aCr en 25–49ml/min y del 26,7% de los que tienen aCr>50ml/min9. Estos mecanismos están presentes y pueden ser identificados antes de la aparición de las manifestaciones clínicas y deberían ser utilizados como marcadores de futuros episodios clínicos.
ENFERMEDAD RENAL Y AFECCIÓN CARDIOVASCULARLa estrecha relación entre la ER y la afección CV se ha comprobado en la población general, en hipertensos, en nefrópatas con enfermedad CV estable preexistente o ICC establecida o tras infarto de miocardio (IM) y en pacientes con microalbuminuria (MA)10.
Filtrado glomerularLos datos del Framingham Heart Study señalaron por primera vez la asociación entre la IR y la morbimortalidad CV11, sobre todo en varones. Más recientemente, Go et al12 comprobaron una relación no lineal entre el FG y el riesgo de muerte, episodios CV y hospitalizaciones, sobre todo a partir de un FG<60ml/min (fig. 1). La mortalidad total asociada a un FG<30ml/min, en ausencia de HD, es muy elevada (un 11,4-14,1% persona/año). La cistatina C, proteína básica procedente de las células nucleadas propuesta como el detector más sensible de la disfunción renal incipiente, puede ser mejor predictor de episodios CV adversos y de mortalidad total que la Cr sérica13, incluso en sujetos sin MA ni reducción del FG14. El estudio CALT ha comprobado una relación directa, tipo dosisrespuesta, entre los valores plasmáticos de cistatina C y la mortalidad total, los episodios CV y la incidencia de ICC, en más de 4.000 pacientes mayores de 65 años15.

Los hipertensos del Hypertension Detection and Follow-up Program Cooperative Group16 con Cr basal≥1,7mg/dl sufrieron más del triple de mortalidad en 8 años, y el RR de morbimortalidad CV fue 1,65 en los pacientes del HOT con FG<60ml/min, calculada utilizando la fórmula de Cockroft-Gault17, y de 1,58 en los de FG>60ml/min18.
En el estudio HOPE, con pacientes con enfermedad vascular y/o DM, con Cr basal<2,3mg/dl y sin ICC, la incidencia acumulada del objetivo primario en 5 años fue del 15% de pacientes con Cr≤1,4mg/dl y del 22% en los de Cr>1,4mg/dl (p<0,001), independientemente de otros FRCV y del grupo de tratamiento19. En pacientes con enfermedad coronaria estable, función sistólica conservada y Cr basal<2mg/dl, se comprobó una relación inversa entre la tasa de eventos y el FG20, y que el bloqueo del sistema renina-angiotensina (SRA) redujo la mortalidad sólo entre los que presentaban un FG<60ml/min/1,73m2.
El FG fue mejor predictor de mortalidad que la clase funcional e incluso que la fracción de eyección (FE) basal en pacientes con ICC avanzada21, y más potente que todos los parámetros analizados, incluida la activación neurohormonal. La IR preexistente ensombrece el pronóstico post-IAM complicado con disfunción sistólica o ICC1. El deterioro del FG predice el desarrollo de ICC tras IM anterior al año22. Los pacientes de alto riesgo que han sobrevivido a un IAM presentan alta prevalencia (aproximadamente un tercio) de IR, que a su vez es un poderoso predictor independiente de nuevos episodios CV, fatales y no fatales. A partir de un FG de 81,0ml/min, cada reducción de 10ml aumenta un 10% el riesgo de muerte o evento no fatal23.
AlbuminuriaLa mortalidad, sobre todo de causa CV, de los pacientes sometidos a HD es superior al 20% anual12, sin logros evidentes a pesar de los esfuerzos realizados.
En el estudio IDNT24, con pacientes hipertensos y diabéticos con albuminuria, no hubo diferencias entre los grupos de tratamiento en cuanto al objetivo CV combinado, pero un análisis posterior25 demostró una relación directa entre la excreción urinaria de albúmina (EUA) al inicio y el objetivo CV. El bloqueo del SRA se asoció a una nefroprotección superior en el mismo tipo de pacientes del estudio RENAAL26, pero sin beneficios significativos en cuanto a la evolución CV, aunque la incidencia de ICC de novo fue menos frecuente en el grupo tratado con losartán. La albuminuria inicial era predictor del objetivo CV combinado, así como de la incidencia de ICC aislada, pero la variación de la EUA en los primeros 6 meses era el mejor predictor evolutivo. Así pues, una reducción del 50% se asoció a un 18% de menor incidencia de episodios CV y a una reducción del riesgo de ICC de un 27%, lo que hace de la albuminuria no sólo un FRCV, sino un objetivo terapéutico o un indicador de la respuesta terapéutica27.
En el estudio de Groningen, que encuestó a más de 40.000 personas, la MA fue un potente predictor de episodios CV en la población general28 a partir de cifras de EUA previamente definidas como normales (EUA<30mg/24h). En los participantes del Third Copenhagen City Heart Study29, una EUA>9μg/min se asoció a un riesgo relativo (RR)=2 para CI y RR=1,9 para muerte al cabo de 5–7 años. Datos del HOPE confirman que la MA es un factor continuo de riesgo incluso a partir de valores tan bajos como 4,4 ^g/min (0,5mg/mmol)30. Cada incremento de 0,4mg/mmol supone un aumento del riesgo de sufrir algún tipo de episodio cardiovascular del 5,9%. Un posterior análisis de los datos del estudio LIFE no sólo relacionó con el riesgo CV la albuminuria basal, sino también el impacto de la reducción en la EUA en la incidencia de episodios31. La relación de la MA con el objetivo combinado del estudio HOPE fue significativo tanto en los diabéticos (RR=1,97) como no diabéticos (RR=1,61)32. La MA fue el más potente predictor de evolución CV adversa en diabéticos, muy superior al tabaquismo, la presión diastólica y el colesterol33.
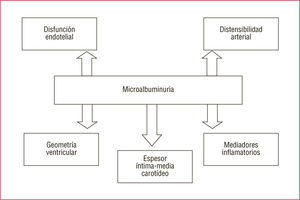
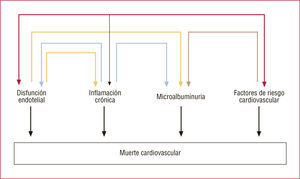
AFECCIÓN CARDIOVASCULAR Y ENFERMEDAD RENAL: MECANISMOS FISIOPATOLÓGICOSDiversos marcadores de morbimortalidad CV coinciden con los de IRT y son HVI, espesor íntima-media carotídeo, rigidez arterial, calcificación aórtica, inflamación y estado procoagulante, disfunción endotelial y estrés oxidativo, anemia, alteraciones de la hemostasis calciofosfórica, albuminuria, proteinuria, homocisteinemia e hiperuricemia.
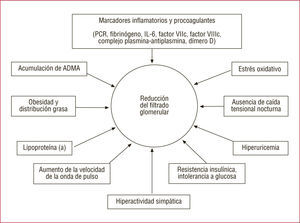
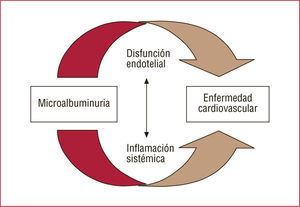
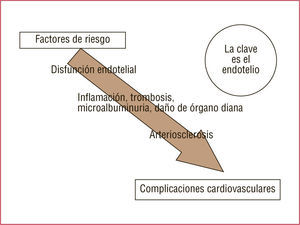
El proceso fisiopatológico que explicaría la asociación de la IRC con el desarrollo de ECV es complejo10,34. Cuando el FG cae por debajo de 60ml/min, se alteran muchas funciones fisiológicas y reguladoras del riñón, como la síntesis de eritropoyetina —con la anemia consiguiente—, y de 1,25-(OH)-vitamina D, que a su vez altera la homeostasis calciofosfórica, aumenta la hormona paratiroidea y favorece la calcificación vascular. La disfunción endotelial y los marcadores inflamatorios y protrombóticos, como la proteína C reactiva (PCR), el fibrinógeno, la interleucina (IL) 6 y el factor VIII, son más prevalentes, así como las concentraciones anormales de lipoproteína (a) (Lp[a]), apolipoproteína (Apo) y homocisteína. Que todos estos factores interactúen entre sí para incrementar el riesgo evolutivo (y en ese caso, de qué forma) no está probado (fig. 2).

La asociación entre IR y afección CV podría explicarse por la elevada prevalencia de los FRCV clásicos, su mayor agresividad para causar lesión de órgano diana y la presencia de anomalías concomitantes que crean un ambiente propicio para la ECV, como disfunción endotelial generalizada, desequilibrio coagulación-fibrinolisis, inflamación y otros FR no tradicionales.
El estudio ARIC35 confirmó la coincidencia de FRCV tradicionales, como el sexo masculino, el tabaquismo, la HTA, la DM y la hipercolesterolemia, con la IRC; pero también otros no tradicionales, como perímetro de la cintura abdominal, apo-B, anemia, hipoalbuminemia e hiperfibrinogenemia. Otros factores como desnutrición, inflamación, anemia, desequilibrio calciofosfórico, homocisteína y fibrinogenemia han generado mucha atención10.
La HTA es un poderoso FR tanto de enfermedad CV como de afección renal, y casi siempre se la halla en la IR. La retención de sodio y la activación del SRA se han considerado máximos factores de la elevación de la presión del nefrópata36. La activación simpática tiene su papel, más complejo desde el descubrimiento de la renalasa, regulador de la función cardiaca y la presión arterial producido por el riñón sano, pero no detectable en pacientes urémicos. La renalasa metaboliza las catecolaminas y parece que se relaciona con la actividad simpática, la HTA y la ER37. La HTA es fundamental en el daño cardiaco de la IRC a través de la HVI38, que se agrava con la reducción de la reserva coronaria y la densidad capilar y que favorece los episodios isquémicos y el deterioro de la función ventricular39,40.
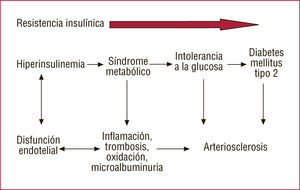
La MA en los hipertensos se asocia a un incremento de 2,5 veces el riesgo de complicaciones CV, como HVI e isquemia miocárdica, así como mayor grosor de la pared carotídea, más afección retiniana y más enfermedad vascular periférica. Los hipertensos con MA muestran alteración del ritmo circadiano de su perfil de presión, con predominio de los hipertensos sin descenso fisiológico nocturno (non dippers); además se comprueba una asociación entre ausencia de la caída de presión nocturna y prevalencia de retinopatía, nefropatía y enfermedad macrovascular. Los predictores más potentes de la presión sistólica nocturna son la MA (p<0,001) y la edad (p<0,01). La prevalencia de HVI es más elevada en los hipertensos sin caída de presión nocturna, así como el espesor del anillo aórtico y el tamaño de la aurícula izquierda41. Los hipertensos con MA presentan mayor RI, con una reducción del 35% en la captación periférica de glucosa y una estrecha correlación entre la EUA y las cifras de glucosa sérica, por lo que la presencia de MA es un potente predictor para el futuro desarrollo de DM42 (figs. 3–5).



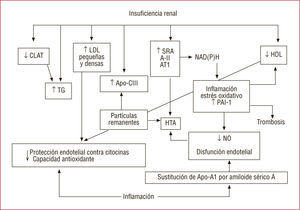
La síntesis hepática de la Apo-AI disminuye y se reducen las concentraciones de lipoproteínas de alta densidad (HDL), importante antioxidante que protege el endotelio de los efectos proinflamatorios de las citocinas. El incremento de la Apo-CIII, inhibidor competitivo de la lipoproteinlipasa, en la IRC enlentece el catabolismo de las lipoproteínas de baja densidad (LDL) y da lugar a acumulaciones de partículas ricas en triglicéridos (TG) en plasma. Las estatinas reducen, aunque modestamente, la proteinuria y enlentecen la progresiva pérdida de funcion renal, lo que confirma el protagonismo de la dislipemia en este proceso43. Los pacientes no diabéticos con IR progresiva muestran concentraciones iniciales de Apo-AIV y TG más elevadas, así como valores de HDL más bajos, que los de función renal estable44.
La MA y la hiperlipemia suelen estar asociadas independientemente de la dieta o el índice de masa corporal (IMC). La albuminuria predice la gravedad de la arteriosclerosis45,46. Los hipertensos con MA muestran valores de TG más elevados, de HDL más bajos y mayor cociente LDL/HDL. Hay correlación positiva entre la Lp(a) y la MA. La relación entre la MA y la dislipemia no se explica por la obesidad o el incremento del IMC, pero puede ser secundaria a la hiperinsulinemia y la RI de los obesos. La MA se asocia con componentes del síndrome metabólico (SM). Hay correlación entre la EUA, la concentración plasmática de insulina en ayunas y la gravedad de la enfermedad coronaria, así como una asociación positiva entre EUA y los valores de TG y Apo-B, e inversa con las de colesterol de las HDL (cHDL). La hiperinsulinemia podría contribuir a la aparición de MA a través de la alteración de la función endotelial. A su vez, la disfunción endotelial podría ser la causa de la MA y de la RI. La MA se asocia a mayor extravasación transcapilar de la albúmina e incremento del factor de Von Willebrand y otros marcadores de disfunción endotelial10 (fig. 6).

El acido úrico (AU) suele estar elevado en pacientes con riesgo CV, sobre todo en mujeres posmenopáusicas47. La reducción del FG disminuye la excreción de AU, por lo que la hiperuricemia suele estar presente en la IRC. La insulina también estimula la reabsorción de sodio y uratos en el túbulo proximal, lo que justifica la hiperuricemia en la RI. Existe una relación continua entre el AU y la presión arterial (sobre todo en jóvenes), y en los animales con daño renal se desarrolla HTA sensible a la sal en relación directa con la concentración de AU. La hiperuricemia también se asocia con la función plaquetaria y la función endotelial. La vida media de las plaquetas de los pacientes con gota, sin enfermedad vascular ni antecedentes CV familiares, es de 2,85 días, mientras que la de los controles sin gota ni enfermedad vascular es de 3,74 días; el recambio plaquetario medio (células/mm/día) es de 58,75 en sujetos con gota y de 42,37 en los controles. La adhesión plaquetaria y la actividad tromboplástica se incrementa de forma proporcional en los gotosos. Todo ello indica aumento de la producción, pero sobre todo destrucción plaquetaria en la gota primaria. El alopurinol, al inhibir la xantina oxidasa, bloquea la formación tanto de AU como de oxidantes, por lo que puede normalizar la formación de NO endotelial disminuida tanto en la DM como en la insuficiencia cardiaca47. Pero la relación del AU con los episodios CV muestra una clara curva en J. De forma especulativa, podría atribuirse el mayor riesgo CV con valores más bajos de AU a la menor actividad antioxidante, mientras que el mayor riesgo con valores más altos estaría en relación con la capacidad del AU de inducir HTA y enfermedad vascular47.
Inflamación, estrés oxidativo y disfunción endotelialLa inflamación es parte de la fisiopatología de la IR y se asocia con todos los principales factores de riesgo de disfunción renal modificables. Los marcadores inflamatorios permanecen elevados en pacientes con IR. La PCR elevada predice la mortalidad total y CV en pacientes en HD, el deterioro renal en sujetos sin nefropatía, así como la HTA, considerada el más importante factor de riesgo de IR. Además, y de forma paralela, la PCR es un predictor independiente de ECV y añade información pronóstica adicional a todos los niveles de riesgo, así como en el síndrome metabólico para cualquier nivel de presión arterial. Del 20 al 65% de los pacientes con IR avanzada muestran altos valores de PCR, por condiciones dependientes de la uremia o de la HD, que predispone a liberación de citocinas proinflamatorias y a otros factores proinflamatorios que generan disfunción endotelial, así como respuestas inflamatorias sistémicas a largo plazo. El 60% de las muertes de pacientes estables en HD son de causa CV, en relación directa con el proceso inflamatorio. Los pacientes con valores basales de PCR más elevados tuvieron un RR 2,4 más alto de muerte y 1,7 de muerte CV (p<0,0001). La PCR elevada fue el mayor predictor de mortalidad junto con la edad y la enfermedad CV previa48.
En sujetos sin DM ni afección renal, los valores de PCR se correlacionan con la reducción evolutiva del FG, así como la hiperfiltración compensadora, signo precoz de deterioro renal. La reducción de la masa de nefronas funcionantes aumenta la filtración de proteínas plasmáticas (proteinuria), y su consecuente reabsorción tubular, que desencadena, por un lado, hipertrofia de las células tubulares por la producción de factor beta de crecimiento transformador y posterior evolución a mioblastos y, por otro, fibrogénesis por expresión de colágeno tipo IV. La reabsorción excesiva de proteínas activa la vía del factor nuclear kappa B, que da lugar a la liberación de sustancias vasoactivas y citocinas proinflamatorias y factores de crecimiento en el intersticio, reclutamiento de células inflamatorias, proliferación de fibroblastos, fibrosis y deterioro progresivo de la función renal. Incluso en etapas precoces de la disfunción renal, hay evidencia de respuesta inflamatoria que parece contribuir al progreso de la enfermedad49. La inflamación interactúa y activa múltiples mecanismos proaterogénicos. Así, la grasa visceral —especialmente— y también —en menor medida— el IMC se relacionan con los marcadores inflamatorios, IL-6, PCR y el factor de necrosis tumoral alfa (TNFa). Bajas cifras de adiponectina, adipocina procedente de la grasa visceral, se asocian con aumento de marcadores inflamatorios plasmáticos, IL-6 y PCR. La adiponectina se relaciona inversamente con la función renal en la DM y se ha comprobado una estrecha relación entre la MA y la adiponectina con la PCR en hipertensos45. La hiperglucemia aguda aumenta la IL-6, el TNFa y la IL-18, y la glucohemoglobina, memoria del control glucémico, se correlaciona estrechamente con los marcadores inflamatorios48,49. El papel de la inflamación en la HTA, el mayor determinante para la progresión de la enfermedad renal, se confirma por su relación tanto con la disfunción endotelial como con la activación del SRA, la sobrerregulación de receptores de tipo I de la angiotensina II (AT1) y, en consecuencia, la vasoconstricción y la proliferación celular. La PCR también se correlaciona con la presión arterial en hipertensos y con el riesgo de HTA para los normotensos, incluso en pacientes sin otros FRCV50.
La expresión de las moléculas de adhesión, ICAM y VCAM, que indica arteriosclerosis precoz, aumenta en pacientes con IR10. Bro et al51 han demostrado un incremento precoz y selectivo del ARNm de ICAM en el endotelio de ratas con aterosclerosis urémica sin cambios en la expresión de selectina ni de VCAM. El principal desencadenante de la respuesta inflamatoria en los pacientes renales es el estrés oxidativo, con la colaboración o al menos complicidad de la PCR al inicio del proceso o en la ampliación de su respuesta. El depósito de PCR en las células endoteliales genera la sobreexpresión de las moléculas de adhesión, la liberación de la proteína quimiotáctica de los monocitos (MCP-1), que atrae los leucocitos para atravesar la barrera endotelial, así como liberación de endotelina 1, potente vasoconstrictor y promotor, por sí mismo, de moléculas de adhesión y de MCP-1 en las células endoteliales, y que cierra así un ominoso círculo. La PCR también puede inhibir la expresión de la sintetasa endotelial de óxido nítrico (NO) (eNOs) y neutralizar el NO producido por el endotelio sano. Al menos in vitro, la PCR regula al alza los receptores AT1, incrementa la proliferación y la migración dependiente de la angiotensina II de las células musculares lisas e incrementa la producción de radicales libres. Se ha comprobado un mayor depósito del complemento C5b9 y PCR en torno a las placas coronarias de pacientes urémicos en comparación con los no urémicos48,49.
La exagerada producción de radicales libres, especies reactivas de oxígeno (ROS), acentúa la disfunción endotelial, la oxidación de las partículas LDL, la expresión de su receptor, LOX-1, y las señales proinflamatorias. El anión superóxido inactiva el NO y da lugar a peroxinitrito. El aumento de las ROS promueve la degradación oxidativa de un cofactor necesario para la sintetasa endotelial de la eNOs, la tetrahidrobiopterina (H4B). El desacoplamiento que se origina da lugar a más producción de anión superóxido en detrimento de la de NO. La actividad de la eNOs también se reduce por aumento de su inhibidor endógeno, que es la dimetilarginina asimétrica (ADMA), favorecido por la inhibición sensible a redox de una enzima, la dimetilarginina dimetilaminohidrolasa (DDAH). La xantina oxidasa y, sobre todo, la nicotinamida adenina dinucleótido fosfato (NADPH) oxidasa constituyen una importante fuente vascular de anión superóxido y oxidan la H4B con lo que se altera la actividad de la eNOs. Se produce así un círculo vicioso, con una producción cada vez mayor de radicales libres y menor disponibilidad de NO. A este desequilibrio contribuye la menor actividad de la superóxido dismutasa (SOD), enzima encargada de la degradación del anión superóxido. El estrés oxidativo desencadena el proceso de lipoperoxidación y formación de las LDL oxidadas, citotóxicas para las células endoteliales que inhiben la respuesta vasodilatadora mediada por NO, y favorecen la agregabilidad plaquetaria. El estrés oxidativo altera el metabolismo celular de modo directo (roturas de ADN, aumento del Ca++ libre intracelular, daño en los transportadores de membrana y alteración de otras proteínas específicas) o indirecto (activación de proteasas secundaria al excesivo aumento de Ca++ libre intracelular, alteración de proteincinasas, síntesis de receptores de membrana, etc.), además de modificar la expresión genética de varias moléculas (eNOs, VCAM-1), así como de la MCP-110,52.
Hay una estrecha relación entre el estrés oxidativo renal y la HTA52. Las ROS pueden aumentar el tono arteriolar aferente, tanto indirectamente (potenciación del feedback tubuloglomerular) como directamente (por reducción de la acción del NO y por vasoconstrictores derivados del endotelio dependientes de la ciclooxigenasa 2, la activación de los receptores del tromboxano y el aumento de la vasorreactividad de las CML). Las consecuencias de la lesión endotelial se amplifican por la exagerada respuesta sistémica del complemento, factor D, sistema CD40 y su ligando, estado protrombótico (factor de Von Willebrand, dímeros) y otros que contribuyen a mayor daño vascular10.
La ADMA, inhibidor selectivo de la eNOs que se asocia a arteriosclerosis avanzada y episodios coronarios, se encuentra elevada en los pacientes con IR, incluso en etapas iniciales, aun en no diabéticos no fumadores, del mismo modo que la homocisteína. El incremento de ADMA presupone disfunción endotelial, con evidente reducción de la vasodilatación dependiente del endotelio. A su vez, la disfunción endotelial favorece el estrés oxidativo, que incrementa la disfunción endotelial, y ambas situaciones limitan la degradación enzimática de la ADMA, lo que cierra un círculo vicioso muy trascendente. La biodisponibilidad de NO está disminuida tanto por síntesis reducida como por exceso de formación de radicales libres (estrés oxidativo)48,50 (figs. 7–9).

H: nicotinamida adenina dinucleótido fosfato oxidasa; PAI-1: inhibidor activador del plasminógeno 1; SRA: sistema reninaangiotensina; TG: triglicéridos.")
Efectos de la insuficiencia renal y la inflamación en las lipoproteínas y la estructura y la función del endotelio. A-II: angiotensina II; Apo-A1: apolipoproteína A1; Apo-CIII: apolipoproteína C-III; AT1: receptor tipo 1 de la angiotensina II; CLAT: colesterol lecitina aciltransferasa; HDL: lipoproteínas de alta densidad; HTA: hipertensión arterial; NAD(P)H: nicotinamida adenina dinucleótido fosfato oxidasa; PAI-1: inhibidor activador del plasminógeno 1; SRA: sistema reninaangiotensina; TG: triglicéridos.
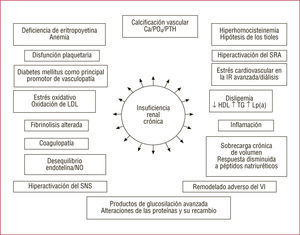
: lipoproteína (a); PTH: paratirina; SNS: sistema nervioso simpático; SRA sistema renina-angiotensina; TG: triglicéridos; VI: ventrículo izquierdo.")
Insuficiencia renal crónica y su impacto en la fisiopatología cardiovascular. HDL: lipoproteínas de alta densidad; LDL: lipoproteínas de baja densidad; Lp(a): lipoproteína (a); PTH: paratirina; SNS: sistema nervioso simpático; SRA sistema renina-angiotensina; TG: triglicéridos; VI: ventrículo izquierdo.
La calcificación vascular precoz, así como de las válvulas cardiacas, es frecuente en la IRC53-55. El 40% de los pacientes con IRC con una tasa media de FG de 33ml/min muestran calcificaciones coronarias, frente al 13% de los controles56. La calcificación afecta a la capa media y suele tratarse de pacientes en HD, incluso antes de iniciarla. Hay una calcificación vascular específica en relación con la HD llamada calcifilaxis o arteriopatía urémica calcificada, que se caracteriza por calcificación difusa de la capa media de las pequeñas y medianas arterias y arteriolas con proliferación intimal y trombosis que pueden causar úlceras, necrosis cutáneas y gangrena de partes acras. Es el resultado de un elevado producto calcio-fosfato y hormona paratiroidea, sin proceso osteogénico activo. Las alteraciones de la homeostasis calcio-fosfato predicen la mortalidad CV en estos pacientes, aunque su mecanismo fisiopatológico no está del todo aclarado57,58, incluso independientemente de la presencia de otros FRCV y del FG59. La calcificación se produce por precipitación pasiva de calcio y fosfatos por excesiva concentración extracelular, promotores de la transformación osteogénica y la formación de hidroxiapatita, así como deficiencia de inhibidores de la calcificación54,55,60. La rigidez aórtica asociada a calcificación favorece la HVI y el aumento de la presión central.
La deficiencia de inhibidores de la calcificación tiene trascendencia clínica. Los bajos niveles de fetuina A en pacientes con IRC se asocian a calcificación vascular acelerada60 y mayor mortalidad61,62; además, el pirofosfato, que previene la formación de cristales de hidroxiapatita y la calcificación, también disminuye en los pacientes en HD63. Por el contrario, la osteoprotegerina aumenta en la IRT64 y se correlaciona con la calcificación vascular y la mortalidad, sobre todo en los que tienen la PCR elevada65. El incremento de leptina, por déficit de excreción, induce calcificación heterotópica a través de sus receptores en el hipotálamo y los receptores betaadrenérgicos osteoblásticos66.
La activación del SRALa activación del SRA tiene lugar en la IRC a través de múltiples mecanismos, en parte como adaptación a la pérdida de masa renal funcionante y los cambios de la hemodinámica renal. La angiotensina II estimula la NADPH oxidasa, que da lugar a anión superóxido y estrés oxidativo, agrava la disfunción endotelial y promueve mediadores inflamatorios como citocinas y quimiocinas, así como factores de crecimiento, moléculas de adhesión, el inhibidor del activador del plasminógeno 1 (PAI 1) y el superóxido barredor de NO. Todo ello contribuye a más disfunción endotelial, remodelado vascular y aterosclerosis progresiva67.
Aunque el riñón produce y secreta renina a la circulación sistémica, es la sobreactivación del SRA local lo que sustancialmente contribuye a la HTA sistémica68. La abundancia de receptores AT1 en el riñón lo hacen muy susceptible a los efectos de la angiotensina II, debido a que las concentraciones de ésta en el espacio intersticial renal y el túbulo proximal son más elevadas que en plasma68. La angiotensina II interactúa con el NO en las CML renales, células mesangiales y la matriz extracelular34.
Los datos clínicos confirman la trascendencia de la activación del SRA en la IRC. Los agentes bloqueadores del SRA han conseguido mayor impacto que otros fármacos en la reducción del riesgo de muerte, infarto de miocardio, ictus, DM y deterioro renal69. Han demostrado beneficios en pacientes con insuficiencia cardiaca, disfunción ventricular, post-IAM, HTA, nefropatía, enfermedad arterial periférica, DM, ictus o accidentes isquémicos transitorios, aunque el beneficio absoluto depende del grado de activación del SRA70.
Ajustándonos al tema objeto de esta revisión, los agentes bloqueadores del SRA reducen la EUA y previenen el desarrollo de MA, marcador de riesgo de enfermedad renal y CV, no solamente en pacientes de alto riesgo con DM, sino también en los de bajo riesgo. En el estudio RENAAL71, el beneficio nefroprotector de losartán puede explicarse casi completamente por su efecto antiproteinúrico. El grado de reducción de la albuminuria se relaciona con el riesgo CV25,72,73, lo que indica que la EUA debe ser utilizada como un objetivo terapéutico similar a la presión arterial para mejorar el pronóstico CV de estos pacientes.
Los resultados del estudio ONTARGET®, presentados en la 57th Annual Scientific Session of the American College of Cardiology, han demostrado que telmisartán es tan efectivo como ramipril en reducir el riesgo de muerte CV, IM, ictus y hospitalizaciones por ICC de los pacientes de alto riesgo, pero sin beneficio aditivo de su combinación.