La miocardiopatía hipertrófica (MCH) es la enfermedad cardiaca hereditaria más frecuente. El desafío actual radica en la clasificación precisa de la patogenicidad de variantes asociadas a las MCH. Para la evaluación inicial de la MCH se recomienda una ecocardiografía transtorácica (ETT). La cardiorresonancia magnética (CRM) también debe considerarse. El objetivo fue revaluar la penetrancia y expresión clínica de la variante patogénica MYBPC3 p.G263*.
MétodosSe estudiaron los principales genes sarcoméricos, mediante next-generation sequencing, en 384 índices con MCH y una cohorte control de 450 individuos sanos. Se identificaron todos portadores de MYBPC3 p.G263* y se realizó cribado familiar. Se recogió información clínica de manera retrospectiva hasta 2015 y prospectiva a partir de entonces. Se realizó un esfuerzo extra para realizar CRM en todos los portadores de la variante independientemente del resultado de la ETT.
ResultadosTrece casos índice con MCH y ninguno de la cohorte control eran portadores de la variante MYBPC3 p.G263*, patogénica según el American College of Medical Genetics and Genomics y la Association for Molecular Pathology. Mediante cribado familiar se identificó a un total de 39 portadores. La mayoría se diagnosticó de MCH asintomática, con inicio tardío de la enfermedad y un curso relativamente benigno, pero con potenciales complicaciones tardías. Se encontró una penetrancia cercana al 70% evaluada por la ETT y del 87% por CRM. La penetrancia era edad-dependiente, y alcanzó el 100% en mayores de 55 años.
ConclusionesMYBPC3 p.G263* comparte con la mayoría de las variantes patogénicas truncantes en este gen un inicio tardío, un curso relativamente benigno en los jóvenes y una alta penetrancia. La CRM podría ser una herramienta útil en la evaluación de portadores independientemente de la ETT.
Palabras clave
La miocardiopatía hipertrófica (MCH) se ha definido como un aumento del grosor de la pared del ventrículo izquierdo que no se explica solo por las condiciones de carga anormales1. La MCH es la cardiopatía hereditaria más frecuente y afecta a 1 de cada 500 personas; tiene un perfil anatomopatológico, clínico y genético bien definido2. Se recomienda realizar en la evaluación inicial una ecocardiografía transtorácica (ETT) a todo paciente con MCH (clase I, nivel B), y debe considerarse la posible conveniencia de una cardiorresonancia magnética (CRM) en la evaluación inicial si los recursos y la experiencia locales lo permiten1.
En la mayoría de los casos, la MCH se hereda en forma de un rasgo genético autosómico dominante3, con una penetrancia incompleta y una expresión clínica diversa1. La secuenciación de los genes de proteínas sarcoméricas identifica una variante causante de la enfermedad en hasta un 60% de los casos3, y la mayor parte de las variantes son de MYH7 (cadena pesada de miosina beta) y MYBPC3 (proteína C transportadora de miosina cardiaca)4. Aunque se recomienda realizar pruebas genéticas a todos los pacientes con MCH para que sea posible el cribado genético de los familiares (clase I, nivel B), la falta de datos sólidos respecto a asociaciones específicas entre genotipo y fenotipo ha reducido su repercusión en el manejo clínico que no va más allá del cribado de detección sistemática1. Dado el bajo número de familias con variantes patogénicas específicas que se ha descrito en la literatura médica, las conclusiones respecto a correlaciones entre genotipo y fenotipo son escasas5. No obstante, esta situación podría mejorar a medida que se obtengan más datos1. Actualmente, el reto real radica en establecer la clasificación exacta de la patogenicidad de las variantes2,6, es necesario dedicarle esfuerzos adicionales. Resulta imprescindible un conocimiento profundo de los mecanismos patogénicos de las variantes que se dan en cada gen para establecer una clasificación fiable. Parece ser que la mayoría de las variantes patogénicas que se dan en los genes sarcoméricos son variantes con cambio de sentido y su comportamiento corresponde a un mecanismo de dominancia negativa5,7. Sin embargo, en el gen MYBPC3, la mayoría de las variantes patogénicas parecen tener un mecanismo patogénico diferente8.
La descripción de variantes patogénicas fundadoras brinda una oportunidad única de evaluar las relaciones clínicas entre un genotipo específico y el fenotipo. Anteriormente hemos descrito la nueva variante MYBPC3 p.G263*, que fue la variante registrada con más frecuencia en una región del norte de España9. El objetivo de este estudio es revaluar la penetrancia y la expresión clínica de esta probable variante patogénica fundadora.
MÉTODOSPoblación del estudio y análisis genéticoSe estudiaron 384 casos índice de MCH de nuestra unidad de referencia de miocardiopatías familiares para determinar los principales genes sarcoméricos mediante next-generation sequencing según lo descrito por Gómez et al.10,11. Además, se secuenció con el mismo panel de genes a 450 individuos de la cohorte sana RENASTUR, de 60-85 años de edad y sin síntomas de MCH, tal como se ha descrito en otra publicación10–13. Se aisló ADN genómico de los pacientes a partir de muestras de sangre. Se obtuvo el consentimiento por escrito de cada uno de los participantes y el protocolo de investigación se atuvo a lo establecido en las directrices éticas de nuestro centro.
Análisis de la clasificación de variantesEl American College of Medical Genetics and Genomics y la Association for Molecular Pathology (ACMG/AMP) recomiendan evaluar la patogenicidad de las variantes para los trastornos mendelianos con una clasificación probabilística («patogénica», «probablemente patogénica», «de significación desconocida = VSD», «probablemente benigna» y «benigna») basándose en múltiples líneas de evidencia6. Se utiliza el criterio de una frecuencia alélica > 5% en el Exome Sequencing Project, el 1000 Genomes Project o el Exome Aggregation Consortium (ExAC) como única base para establecer una interpretación de benignidad. En consecuencia, las variantes de menor frecuencia descritas tras la aparición de la guía de ACMG/AMP de 2015, como la MYBPC3 p.G263*, se evaluaron basándose en estos documentos6.
Evaluación clínicaSe identificó y trató a los portadores de MYBPC3 p.G263* según lo indicado en las guías de la MCH1. Se elaboró un árbol genealógico de cada probando y se realizó un cribado genético de todos los familiares a los que se pudo acceder.
Se realizó una evaluación clínica que incluyó los antecedentes personales familiares, una exploración física, electrocardiograma de 12 derivaciones en reposo, monitorización Holter de 24 h, ETT y CRM si era posible. La información clínica se registró retrospectivamente antes de 2015 y prospectivamente desde 2015 hasta la fecha.
Dos pacientes fallecieron sin que se les hubiera practicado una CRM. Fue necesario el esfuerzo adicional de realizar una CRM a todos los portadores de MYBPC3 p.G263* vivos, con independencia de los resultados de la ETT previa. La pasaron todos los individuos excepto los que tenían claustrofobia o un desfibrilador automático implantable, las embarazadas y los individuos de edad avanzada. El diagnóstico clínico de MCH se estableció por el aumento del grosor de la pared del ventrículo izquierdo (≥ 15 mm o ≥ 13 mm en los familiares de primer grado con MCH), medido con alguna de las técnicas de imagen, que no se explicara solo por las condiciones de carga anormales1. Se utilizó la calculadora del riesgo de muerte súbita por MCH14.
Análisis estadísticoSe utilizó el software estadístico SPSS v.19. Los valores se expresan en forma de media ± desviación estándar para las variables continuas y en forma de frecuencia y porcentaje para las variables discretas. Se utilizó la prueba de la χ2 o la prueba exacta de Fisher para comparar las frecuencias, mientras que las diferencias en las variables continuas se evaluaron con la prueba de la t de Student o la prueba de la U de Mann-Whitney. Se consideró significativo un valor de p < 0,05.
RESULTADOSNext-generation sequencingSe identificó la presencia de una variante patogénica/probablemente patogénica en un 34% de los casos índice de MCH (132/384), y el gen afectado con más frecuencia fue MYBPC3 (79/384 [21%]).
Se identificó la variante MYPBC3 p.G263* en 13 casos índice de la cohorte de MCH (13/384) aparentemente no relacionados entre sí (figura 1). La variante MYPBC3 p.G263* supone un 10% del total de variantes patogénicas/probablemente patogénicas (13/132) halladas en esta cohorte y un 16,5% de las del gen MYBPC3 (13/79). No se identificaron otras variantes patogénicas/probablemente patogénicas en estos 13 pacientes (en un artículo previo se presentó la variante probablemente benigna MYBPC3 R326Q, y la VSD MYH7 A100T)9.

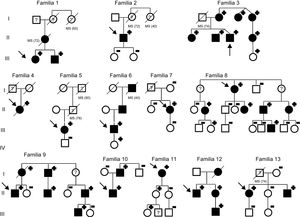
Árbol genealógico de familias con miocardiopatía hipertrófica portadoras de la variante MYBPC3 p.G263*. MS: muerte súbita. Los símbolos indican el sexo y el estado respecto a la enfermedad: +, portadores; –, no portadores; ?, fenotipo desconocido; recuadro, sexo masculino; círculo, sexo femenino; negro, fenotipo de miocardiopatía hipertrófica; rayado, fallecido; blanco, no afectado; sin signo, no estudiado.
Ninguno de los 450 controles era portador de la MYBPC3 p.G263*.
Clasificación de las variantesSegún la clasificación de ACMG/AMP6, la MYBPC3 p.G263* es una variante patogénica. Se trata de una variante de truncación en un gen en el que la pérdida de función constituye un mecanismo de enfermedad conocido, y la evidencia computacional confirma un efecto deletéreo en el gen. Además, no está presente en los controles de las bases de Exome Sequencing Project, 1000 Genomes Project y ExAC ni en nuestra cohorte de control del RENASTUR. Además, la sólida información de segregación obtenida en múltiples familiares afectados (figura 1) respalda claramente su patogenicidad.
Población del estudioLa MYPBC3 p.G263* se identificó en 13 casos índice (media de edad, 56 ± 14,7 años; 6 varones). Se evaluó a un total de 66 individuos de estas 13 familias (figura 1). Las pruebas genéticas de los familiares identificaron a 39 portadores (17 varones) y 27 no portadores.
Un total de 34 portadores (el 87,2%; el 50% varones) cumplían los criterios de la MCH (tabla 1). La penetrancia de la enfermedad, evaluada por ETT y CRM, mostró una dependencia de la edad, de manera que se alcanzaba una penetrancia del 96,7% en los portadores de más de 40 años y del 100% en los de más de 55.
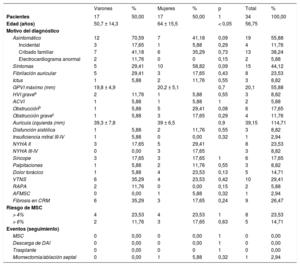
Características de los 34 portadores de la variante MYBPC3 p.G263X afectados por una miocardiopatía hipertrófica
| Varones | % | Mujeres | % | p | Total | % | |
|---|---|---|---|---|---|---|---|
| Pacientes | 17 | 50,00 | 17 | 50,00 | 1 | 34 | 100,00 |
| Edad (años) | 50,7 ± 14,3 | 64 ± 15,5 | < 0,05 | 56,75 | |||
| Motivo del diagnóstico | |||||||
| Asintomático | 12 | 70,59 | 7 | 41,18 | 0,09 | 19 | 55,88 |
| Incidental | 3 | 17,65 | 1 | 5,88 | 0,29 | 4 | 11,76 |
| Cribado familiar | 7 | 41,18 | 6 | 35,29 | 0,73 | 13 | 38,24 |
| Electrocardiograma anormal | 2 | 11,76 | 0 | 0 | 0,15 | 2 | 5,88 |
| Síntomas | 5 | 29,41 | 10 | 58,82 | 0,09 | 15 | 44,12 |
| Fibrilación auricular | 5 | 29,41 | 3 | 17,65 | 0,43 | 8 | 23,53 |
| Ictus | 1 | 5,88 | 2 | 11,76 | 0,55 | 3 | 8,82 |
| GPVI máximo (mm) | 19,8 ± 4,9 | 20,2 ± 5,1 | 0,7 | 20,1 | 55,88 | ||
| HVI gravea | 2 | 11,76 | 1 | 5,88 | 0,55 | 3 | 8,82 |
| ACVI | 1 | 5,88 | 1 | 5,88 | 1 | 2 | 5,88 |
| Obstrucciónb | 1 | 5,88 | 5 | 29,41 | 0,08 | 6 | 17,65 |
| Obstrucción gravec | 1 | 5,88 | 3 | 17,65 | 0,29 | 4 | 11,76 |
| Aurícula izquierda (mm) | 39,3 ± 7,8 | 39 ± 6,5 | 0,9 | 39,15 | 114,71 | ||
| Disfunción sistólica | 1 | 5,88 | 2 | 11,76 | 0,55 | 3 | 8,82 |
| Insuficiencia mitral III-IV | 1 | 5,88 | 0 | 0,00 | 0,32 | 1 | 2,94 |
| NYHA II | 3 | 17,65 | 5 | 29,41 | 8 | 23,53 | |
| NYHA III-IV | 0 | 0,00 | 3 | 17,65 | 3 | 8,82 | |
| Síncope | 3 | 17,65 | 3 | 17,65 | 1 | 6 | 17,65 |
| Palpitaciones | 1 | 5,88 | 2 | 11,76 | 0,55 | 3 | 8,82 |
| Dolor torácico | 1 | 5,88 | 4 | 23,53 | 0,13 | 5 | 14,71 |
| VTNS | 6 | 35,29 | 4 | 23,53 | 0,42 | 10 | 29,41 |
| RAPA | 2 | 11,76 | 0 | 0,00 | 0,15 | 2 | 5,88 |
| AFMSC | 0 | 0,00 | 1 | 5,88 | 0,32 | 1 | 2,94 |
| Fibrosis en CRM | 6 | 35,29 | 3 | 17,65 | 0,24 | 9 | 26,47 |
| Riesgo de MSC | |||||||
| > 4% | 4 | 23,53 | 4 | 23,53 | 1 | 8 | 23,53 |
| > 6% | 2 | 11,76 | 3 | 17,65 | 0,63 | 5 | 14,71 |
| Eventos (seguimiento) | |||||||
| MSC | 0 | 0,00 | 0 | 0,00 | 1 | 0 | 0,00 |
| Descarga de DAI | 0 | 0,00 | 0 | 0,00 | 1 | 0 | 0,00 |
| Trasplante | 0 | 0,00 | 0 | 0 | 1 | 0 | 0,00 |
| Miomectomía/ablación septal | 0 | 0,00 | 1 | 5,88 | 0,32 | 1 | 2,94 |
ACVI: ausencia de compactación del ventrículo izquierdo; AFMSC: antecedentes familiares de muerte súbita cardiaca; CRM: cardiorresonancia magnética; DAI: desfibrilador automático implantable; GPVI: grosor de la pared ventricular izquierda; HVI: hipertrofia ventricular izquierda; MSC: muerte súbita cardiaca; NYHA: New York Heart Association; RAPA: respuesta anormal de la presión arterial al ejercicio en posición erguida; VTNS: taquicardia ventricular no sostenida en monitorización Holter.
Salvo otra indicación, los valores expresan n (%) o media ± desviación estándar.
De los 39 portadores de MYBPC3 p.G263*, 24 fueron examinados mediante CRM, con independencia de las exploraciones previas de ETT. Se observó fibrosis en un 37,5% de ellos. La CRM de 2 portadores con MCH mostró unas características compatibles con ausencia de compactación del ventrículo izquierdo (ACVI). La CRM identificó criptas no apreciadas en la ETT en 2 portadores sin MCH.
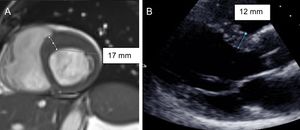
La RM fue la clave para diagnosticar adecuadamente a 8 pacientes (figura 2). Sin la CRM, la hipertrofia ventricular izquierda de 6 portadores habría pasado inadvertida con la ETT. Además, la CRM facilitó la evaluación de una hipertrofia limítrofe identificada en la ETT y un diagnóstico diferencial entre la MCH y la miocardiopatía hipertensiva. La media de tiempo entre la ETT y la CRM fue 1,2 ± 1,25 años. Si no se hubiera realizado la CRM, las tasas de penetrancia evaluadas solo con la ETT se habrían reducido a un 66,7-71,8%. La CRM no identificó ningún resultado falso positivo de la ETT.
 y que se visualiza en la cardiorresonancia magnética (B).")
Los varones con el fenotipo de MCH tenían una edad significativamente inferior que las mujeres. Tiene interés señalar que todos los portadores sin afección clínica fueron mujeres de menos de 55 años. Estos resultados corresponden a una penetrancia del 100% de los varones y el 77,3% de las mujeres. Aunque hubo ligeras diferencias entre sexos por lo que respecta a las características clínicas (tabla 1), no alcanzaron la significación estadística. La media de edad de los varones en el momento del diagnóstico fue 50 ± 14 años y la de las mujeres, 59 ± 15,5. La media de edad de los portadores varones en el momento de realizar las pruebas genéticas fue 45 ± 15 años; la de las mujeres portadoras, 54 ± 19 y la de las portadoras con MCH, 61 ± 15.
La mayoría de los pacientes (55,9%) estaban asintomáticos en el momento del diagnóstico, sobre todo en el contexto del cribado familiar (tabla 1). Aunque los resultados electrocardiográficos no fueron el principal motivo para la consulta cardiológica, en un 85% de los portadores estos resultados fueron anormales.
Solo 1 paciente tenía antecedentes familiares de muerte súbita cardiaca (MSC) considerando a los familiares de primer grado de menos de 40 años (o de cualquier edad si se había diagnosticado una MCH). No obstante, la MSC podría darse a edades avanzadas y también en familiares de segundo grado (figura 1).
El riesgo medio de MSC fue bajo. Sin embargo, en 9 pacientes el riesgo fue superior (4 con riesgo intermedio y 5 con riesgo alto). Un paciente con un riesgo elevado falleció a causa de una insuficiencia cardiaca antes de implantársele un desfibrilador automático implantable. Se implantó un desfibrilador automático implantable a los otros 3 pacientes con alto riesgo y el quinto lo tiene pendiente. En 3 de los 4 pacientes con riesgo intermedio se optó también por el uso de un desfibrilador automático implantable y se está considerando la posible conveniencia del implante para el cuarto. No hubo ningún caso de MSC ni de descargas de un desfibrilador automático implantable. El riesgo de uno de los pacientes ha disminuido a menos de un 4% a causa de la edad.
Solo 3 pacientes presentaron deterioro de la función sistólica del ventrículo izquierdo (de carácter moderado/grave en los 2 con ACVI) y 3 tuvieron ictus (1 de ellos con una fibrilación auricular como factor de riesgo, otro con una ACVI y el tercero con fibrilación auricular y ACVI).
DISCUSIÓNClasificación exacta de las variantesLa creciente concienciación pública respecto a las enfermedades hereditarias y las nuevas técnicas de secuenciación disponibles han aumentado drásticamente la demanda clínica de pruebas genéticas, así como el número de variantes que requieren una interpretación bioinformática y clínica2. Actualmente, el número de variantes patogénicas de la MCH descritas es superior al esperado. Walsh et al.15 analizaron los datos de 7.855 individuos a los que realizaron pruebas para la identificación de miocardiopatías hereditarias en un total de 60.706 muestras de referencia de ExAC. Se observó que un 11,7% de los individuos del ExAC presentan variantes de MCH descritas, lo cual supone un enorme exceso de prevalencia de la enfermedad, que es incompatible con la prevalencia real de la MCH (0,5%). Esto demostró que muchas de las variantes descritas se han clasificado de manera errónea. Por este motivo, debe ponerse mayor empeño en clasificar adecuadamente la patogenicidad de las variantes. Es esencial disponer de un conocimiento profundo de los mecanismos de patogenicidad de cada gen y los criterios de ACMG/AMP6 deben analizarse con precaución.
Las variantes causantes de miocardiopatía en la mayoría de las proteínas de los miofilamentos se incorporan en la sarcómera y actúan como negativos dominantes, como las variantes de sentido erróneo en el gen MYH75,7. En cambio, la mayoría de las variantes patogénicas del gen MYBPC3 causan truncación y la MCH a través de la haploinsuficiencia8,16,17. Walsh et al.15 confirmaron la asociación entre la MCH y la variante con cambio de sentido en el gen MYH7 y la variante truncada en el gen MYBPC316. Estos autores analizaron la predicción de variantes truncadas y no truncadas en las miocardiopatías y calcularon su fracción etiológica, que es una estimación de la proporción de casos en que la presencia de una variante rara en un gen causa enfermedad. En los genes en que los alelos causantes de truncación causan la enfermedad, la odds ratio para la comparación de los portadores de variantes raras con los no portadores es habitualmente superior. Las variantes no truncadas del gen MYBPC3 mostraron una odds ratio de 5,7 (fracción etiológica de 0,8), mientras que las variantes truncadas tuvieron una odds ratio casi 21 veces superior (fracción etiológica, 0,99). En cambio, las variantes truncadas en MYH7 tuvieron una odds ratio de solo 1,7 (fracción etiológica, 0,4) y las variantes no truncadas, 12 (fracción etiológica, 0,92)15.
Debe aportarse información sobre las variantes patogénicas lo bastante caracterizadas para mejorar las bases de datos mundiales y facilitar su interpretación en otras zonas del mundo donde pudieran no estar presentes.
Fenotipo clínico y comparación con otras variantes patogénicas de MYBPC3Aunque las variantes patogénicas de MYBPC3 se han asociado con un inicio tardío de la MCH (edad > 40 años)18, también se ha descrito hipertrofia leve, una incidencia baja de MSC, un curso clínico relativamente benigno y una variabilidad del inicio y el pronóstico de la enfermedad19,20. No obstante, no se han hallado diferencias de fenotipo clínico atribuibles a un tipo específico de una variante de MYBPC321. La alta prevalencia de variantes patogénicas de tipo fundador brinda la oportunidad de definir sus perfiles clínicos. Se han identificado muchas variantes patogénicas fundadoras en MYBPC3 en algunas poblaciones; tiene interés señalar que todas ellas son variantes truncadas17 y constituyen una gran parte (de un 10-25% a un 58%) de las variantes patogénicas detectadas en sus países de origen22–29. El inicio tardío de las complicaciones de la MCH con peligro para la vida, con una aparición más allá de la edad reproductiva, ha permitido su transmisión a las generaciones siguientes24,25.
La CRM permite realizar una evaluación tomográfica tridimensional de la anatomía cardiaca y se ha convertido en un excelente instrumento para evaluar las miocardiopatías. De hecho, se considera que es el patrón de referencia para la evaluación no invasiva de la masa, los volúmenes y la fracción de eyección ventriculares. La CRM es superior a la ETT en la detección de la hipertrofia ventricular izquierda apical y anterolateral30 y de los trombos31. En algunas series, la CRM ha identificado criptas miocárdicas en hasta un 70% de los portadores de variantes patogénicas sin MCH32–34. Aunque se recomienda el uso de la CRM para pacientes con ventanas ecocardiográficas inadecuadas (IB), a veces su papel en la fase de cribado para la detección de la MCH es limitado, debido a la escasez de recursos locales1. Así pues, la penetrancia de las variantes patogénicas en la MCH se evalúa con frecuencia mediante las exploraciones de ETT.
Lamentablemente, en muchos estudios de variantes patogénicas fundadoras, como el de Adalsteinsdottir et al.22, solo se incluyó a probandos o pacientes con un fenotipo de MCH conocido, por lo que no fue posible evaluar la penetrancia de la enfermedad. La variante fundadora descrita en el gen MYBPC3 (transición c.927-2ANG en el intrón 11) es la principal causa (58%) de MCH en Islandia22. La variante MYBPC3 Gln1061X supone un 17% del total de casos de Finlandia26 y se asocia con fenotipos benignos o intermedios. Otra variante fundadora patogénica de Italia, la MYBPC3 p.F305Pfs*27, supone alrededor de un 20% de los casos de MCH de su cohorte24. Los pacientes portadores de esta variante muestran también un inicio tardío de la MCH y MSC después de la cuarta década de la vida.
El estudio más amplio realizado hasta la fecha, en el que se compararon las características fenotípicas observadas en la CRM de 125 pacientes con MCH portadores de variantes patogénicas en MYH7 (53 pacientes) y MYBPC3 (75 pacientes), no observó diferencias fenotípicas entre los 2 grupos35. Lamentablemente, una vez más este estudio se realizó en pacientes con un fenotipo de MCH ya conocido y no fue posible evaluar las diferencias de penetrancia entre los pacientes portadores de variantes patogénicas en MYH7 y MYBPC3. Sin embargo, los autores sí observaron diferencias en los pacientes de edad > 40 años: los portadores de variantes en MYBPC3 presentaban una fracción de eyección del ventrículo izquierdo significativamente menor.
En Japón, Kubo et al.28 describieron una penetrancia del 76,9% en sus 39 portadores de MYBPC3 V592fs/8, si bien el estudio se basó en exploraciones de ETT solamente (tabla 2). La penetrancia de la enfermedad fue del 100% de los participantes de edad > 50 años y las complicaciones como la MSC y la disfunción sistólica del ventrículo izquierdo con insuficiencia cardiaca se dieron también después de los 50 años. La variante MYBPC3 Val592fs/8 se identificó en un 16% de los probandos con MCH28.
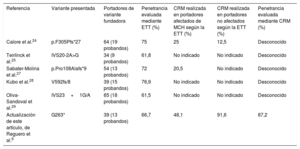
Comparación de las variantes patogénicas fundadoras
| Referencia | Variante presentada | Portadores de variante fundadora | Penetrancia evaluada mediante ETT (%) | CRM realizada en portadores afectados de MCH según la ETT (%) | CRM realizada en portadores no afectados según la ETT (%) | Penetrancia evaluada mediante CRM (%) |
|---|---|---|---|---|---|---|
| Calore et al.24 | p.F305Pfs*27 | 64 (19 probandos) | 75 | 25 | 12,5 | Desconocido |
| Teirlinck et al.25 | IVS20-2A>G | 34 (9 probandos) | 61,8 | No indicado | No indicado | Desconocido |
| Sabater-Molina et al.27 | p.Pro108Alafs*9 | 54 (13 probandos) | 72 | 20,5 | No indicado | Desconocido |
| Kubo et al.28 | V592fs/8 | 39 (15 probandos) | 76,9 | No indicado | No indicado | Desconocido |
| Oliva-Sandoval et al.29 | IVS23+1G/A | 65 (18 probandos) | 61,5 | No indicado | No indicado | Desconocido |
| Actualización de este artículo, de Reguero et al.9 | G263* | 39 (13 probandos) | 66,7 | 48,1 | 91,6 | 87,2 |
CRM: cardiorresonancia magnética; ETT: ecocardiografía transtorácica; MCH: miocardiopatía hipertrófica.
Teirlinck et al.25 describieron una penetrancia de la MCH del 61,8% en 34 portadores de la MYBPC3 IVS20-2A>G, basándose en las exploraciones de ETT sin CRM (tabla 2). Tiene interés señalar que el diagnóstico se hizo en una fase de la vida posterior a la de los portadores con otras variantes patogénicas en los demás genes, pese a haber presentado los primeros síntomas a la misma edad. Cabe presumir que el retraso diagnóstico se debió a una aparición tardía de la hipertrofia ventricular25.
En un estudio de otra variante en España, basado en exploraciones de ETT realizadas a 65 portadores de MYBPC3 IVS23+1G/A, se observó una penetrancia del 61,5% (tabla 2)29.
En estudios recientes se está empezando a incluir la CRM en los análisis. Se ha descrito una penetrancia del 72% en la nueva variante MYBPC3 p.Pro108Alafs*9 observada en España (tabla 2)27. Sin embargo, en esta cohorte, solo se realizó la CRM a 8 pacientes (el 14,8% de los portadores)27.
En los 64 portadores de la variante de truncación MYBPC3 p.F305Pfs*2, la penetrancia de la enfermedad fue del 75% (tabla 2). No obstante, la CRM se realizó solo a 12 portadores afectados (el 25% de los portadores) y 2 portadores no afectados24.
La variante MYBPC3 p.G263* tiene en común con otras variantes truncadas patogénicas en MYBPC3 la alta penetrancia, con un perfil de riesgo de MSC bajo y un curso clínico relativamente benigno, según se ha descrito anteriormente9. Este estudio aporta una actualización clínica con un seguimiento de 5 años, así como nueva información sobre los familiares y un aumento de los diagnósticos con la CRM. En esta variante patogénica, hubo también complicaciones de MCH de inicio tardío y con peligro para la vida, pero a una edad avanzada, especialmente después de los 40 años, lo cual respalda el perfil a priori benigno de esta variante patogénica. De hecho, muchos de los pacientes estaban asintomáticos en el momento del diagnóstico (debido a un cribado familiar o como hallazgo incidental). No se ha demostrado que haya un efecto de fundador, ni fue este el objetivo del estudio. Sin embargo, tanto su frecuencia elevada (el 10% del total de variantes patogénicas/probablemente patogénicas en la MCH) como el aislamiento histórico y geográfico de nuestra región respaldan claramente un efecto de fundador de la variante.
No todos los genes relacionados con la MCH con variantes patogénicas ni todas las variantes patogénicas del mismo gen se comportan de la misma manera en cuanto a forma de presentación clínica y evolución36. Sin embargo, en general, las variantes truncadas patogénicas en MYBPC3 parecen comportarse de manera similar, sea cual sea la variante específica de que se trate. Dado su inicio tardío, su curso clínico es, en general, benigno. Sin embargo, los clínicos deben ser conscientes de las posibles complicaciones tardías con peligro para la vida (p. ej., MSC, ictus, disfunción sistólica del ventrículo izquierdo e insuficiencia cardiaca), en especial a partir de los 40-50 años, a diferencia de las variantes patogénicas de otros genes relacionados con la MCH, en las que el «periodo de riesgo» habría pasado ya.
Además, en nuestra cohorte, la CRM facilitó la identificación del fenotipo de MCH. Sin CRM, las tasas de penetrancia serían similares a las descritas en las demás variantes del gen MYPBC3 (tabla 2). De hecho, la primera vez que se identificó la variante MYBPC3 p.G263* en nuestra población, se había descrito una penetrancia aparentemente baja9. Esta nueva evaluación ha permitido actualizar su penetrancia y su expresión clínica, identificando a pacientes con MCH que de otro modo habrían pasado inadvertidos. Estos datos nos llevan a preguntarnos si, en realidad, la penetrancia de otras series puede haberse subestimado también, sobre todo en los pacientes jóvenes. Tal como ya indicaron Valente et al.37, hay un porcentaje de individuos que pueden estar afectados de MCH y pasar inadvertidos sin el apoyo de la CRM. En su estudio, la CRM identificó una hipertrofia leve en alrededor de un 10% de los portadores de variantes patogénicas con un grosor de la pared normal en la ETT. En consecuencia, en el cribado familiar de portadores de variantes patogénicas, cuando la ETT sea normal o las imágenes no sean óptimas, los resultados del cribado pueden aumentarse con la exploración de CRM.
Además, en nuestra población, la CRM identificó a 2 pacientes cuya MCH coexistía con unas manifestaciones estructurales compatibles con una ACVI. En consecuencia, esta variante patogénica es un ejemplo de la heterogeneidad alélica, con una expresión fenotípica variable en los portadores con distintas miocardiopatías (MCH y ACVI), lo cual plantea de nuevo la cuestión de si se trata en realidad de manifestaciones diferentes del mismo espectro de miocardiopatías38. El hecho de que 2 de los pacientes de nuestra población con ACVI tuvieran un ictus y disfunción sistólica moderada/grave, que son las 2 complicaciones más graves de esta cohorte, hace que esta cuestión resulte intrigante.
LimitacionesAlgunos de los familiares rechazaron que se les practicara un estudio clínico o genético. Además, en el análisis de imagen no se utilizó enmascaramiento, puesto que en las solicitudes de CRM y de ETT se especificaba «examen de detección de la MCH familiar». Dado que el tiempo transcurrido entre la ETT y la CRM no fue homogéneo y fue demasiado largo en algunos casos, hay que tener precaución al interpretar estos datos. Por último, en este estudio se presenta información acerca de la penetrancia en función de la edad. Sin embargo, muchos de los pacientes afectados estaban asintomáticos y fueron diagnosticados por casualidad. En consecuencia, la edad al primer contacto con un cardiólogo no fue homogénea ni se pudo realizar exploraciones de imagen estandarizadas (a la misma edad y con la misma periodicidad). Así pues, no fue posible asumir otras conclusiones respecto a los datos de dependencia de la edad en esta población.
CONCLUSIONESLa variante patogénica de MYBPC3 p.G263* tiene en común con la mayoría de las variantes truncadas patogénicas de este gen las 3 características siguientes: un inicio tardío, un curso clínico relativamente benigno en los jóvenes y una penetrancia elevada dependiente de la edad. Para identificar a todos los pacientes afectados, la CRM podría ser un instrumento útil de evaluación de los individuos que son portadores pese a una ETT aparentemente normal.
CONFLICTO DE INTERESESNo se declara ninguno.
- –
El reto actual en la MCH, que es la cardiopatía hereditaria más frecuente, radica en la clasificación exacta de la patogenicidad de las diversas variantes.
- –
La falta de datos firmes respecto a asociaciones específicas entre genotipo y fenotipo ha reducido su repercusión en el manejo clínico, que no va más allá del cribado de detección sistemática.
- –
Se ha analizado la variante patogénica MYBPC3 p.G263* y se ha realizado una revisión de la literatura médica sobre las variantes patogénicas fundadoras, para buscar posibles correlaciones entre genotipo y fenotipo. Esta variante tiene en común con la mayoría de las variantes truncadas patogénicas en este gen el inicio tardío y el curso clínico relativamente benigno en los individuos jóvenes, así como una gran penetrancia dependiente de la edad. Además, la penetrancia podría ser mayor aún si se evaluara mediante CRM.