
La miocardiopatía hipertrófica (MCH) es una enfermedad con expresión variable, causada principalmente por mutaciones en genes sarcoméricos, aunque otros factores podrían estar modulando el fenotipo. El objetivo es determinar si el sexo, la hipertensión arterial o la actividad física son moduladores de la gravedad de la enfermedad y establecer su papel en la penetrancia en relación con la edad al diagnóstico de la MCH.
MétodosSe evaluó a 272 individuos (media de edad, 49 ± 17 años; el 57% varones) procedentes de 72 familias con mutación causal y se estudió la relación del sexo, la hipertensión y la actividad física con la hipertrofia ventricular izquierda.
ResultadosLa proporción de afectados aumenta con la edad. Se diagnosticó a los varones una mediana ajustada de 12,5 años (IC95%, –17,52 a –6,48; p < 0,001) antes que a las mujeres. A los pacientes hipertensos, se les diagnosticó MCH una mediana ajustada de 10,8 años (IC95%, 6,28-17,09; p < 0,001) más tarde que a los normotensos. A los individuos que hacían ejercicio, se los diagnosticó significativamente antes (mediana ajustada, 7,3 años; IC95%, –14,49 a –1,51; p = 0,016). El sexo, la hipertensión y el ejercicio no resultaron significativamente asociados con la gravedad de la hipertrofia del ventrículo izquierdo. Los factores explorados no influyen en la supervivencia ajustada libre de muerte súbita y el evento combinado.
ConclusionesSe diagnostica a los varones y los deportistas portadores de mutaciones sarcoméricas antes que a las mujeres y los individuos sedentarios. Los portadores de mutaciones sarcoméricas hipertensos tienen un retraso en el diagnóstico. El sexo, la hipertensión y el ejercicio no se asocian con la gravedad de la enfermedad en portadores de mutaciones causales de MCH.
Palabras clave

La miocardiopatía hipertrófica (MCH) es una enfermedad cardiaca genética caracterizada por su heterogeneidad morfológica, funcional, clínica y pronóstica1–7. La expresión fenotípica variable y su penetrancia incompleta8 han constituido un obstáculo para poder conocer plenamente el espectro clínico y las consecuencias de la enfermedad. La causa de la MCH son principalmente las mutaciones en genes que codifican las proteínas contráctiles sarcoméricas9,10
Se han identificado mutaciones patógenas en más de 12 genes11. Las mutaciones de los genes MYH7 y MYBPC3 son la causa de alrededor del 80% de los casos en que se ha determinado el genotipo12,13.
Aunque en la guía de práctica clínica sobre MCH14 no se recomienda el ejercicio intenso y de competición, la asociación entre la actividad física y los eventos en la MCH es débil. No obstante, se ha aceptado que la actividad física es un desencadenante de arritmias ventriculares y muerte súbita (MS) en pacientes con MCH15. Los clínicos valoran y asesoran a pacientes con MCH, o incluso a portadores no afectados, que solicitan asesoramiento respecto a la actividad física. Es crucial esclarecer si las variaciones en el estilo de vida pueden modificar la penetrancia de la enfermedad16.
Se ha demostrado que el sexo del paciente es una variable importante en la evolución natural de la caracterización y el tratamiento de diversos trastornos cardiovasculares adquiridos17–22.
De manera similar a lo que ocurre con la actividad física intensa, se cree que la hipertensión (HT) aumenta el grado de hipertrofia del ventrículo izquierdo (HVI) a través de la inducción de una hipertrofia miocardiocitaria23.
Las observaciones realizadas de diferencias intrafamiliares no se explican por la heterogeneidad mutacional y, por consiguiente, deben intervenir en ello factores ambientales21.
El objetivo principal de este estudio es comparar la penetrancia, entendida como edad al diagnóstico de la MCH, las características clínicas y la evolución clínica en una cohorte amplia de familias con MCH genotipificada con mutaciones en MYBPC3 y MHY7, por lo que respecta al sexo del paciente, la presencia o ausencia de HT y la intensidad de la actividad física.
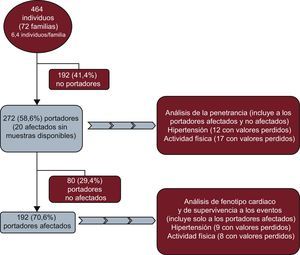
MÉTODOSMuestra del estudioSe seleccionó a todos los participantes de un conjunto inicial de 72 familias (464 individuos) con MCH. De estas 72 familias, se excluyó a 192 (41,4%) familiares normales que no eran portadores de la mutación familiar. En la muestra de 272 portadores de 72 familias diferentes, se incluyó como portadores obligados a 20 individuos afectados con variables clínicas pero sin muestras disponibles (figura 1).

Formaron nuestra cohorte total 272 individuos (media de edad, 49,0±17,5 años; el 57% varones). Un total de 221 (81,3%) eran portadores de una mutación causal en el gen MYBPC3 y 51 (18,8%) en MYH7 (tabla 1).
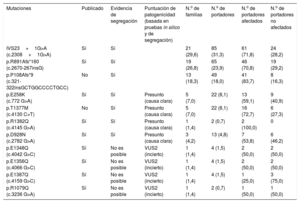
Frecuencia de mutaciones en nuestros portadores de MYH7-MYBPC3
| Mutaciones | Publicado | Evidencia de segregación | Puntuación de patogenicidad (basada en pruebas in silico y de segregación) | N.o de familias | N.o de portadores | N.o de portadores afectados | N.o de portadores no afectados |
|---|---|---|---|---|---|---|---|
| IVS23+1G>A (c.2308+1G>A) | Sí | Sí | 21 (29,6) | 85 (31,3) | 61 (71,8) | 24 (28,2) | |
| p.R891Afs*160 (c.2670-267insG) | Sí | Sí | 19 (26,8) | 65 (23,9) | 46 (70,8) | 19 (29,2) | |
| p.P108Afs*9 (c.321-322insGCTGGCCCCTGCC) | No | Sí | 13 (18,3) | 49 (18,0) | 41 (83,7) | 8 (16,3) | |
| p.E258K (c.772 G>A) | Sí | Sí | Presunto (causa clara) | 5 (7,0) | 22 (8,1) | 13 (59,1) | 9 (40,9) |
| p.T1377M (c.4130 C>T) | No | Sí | Presunto (causa clara) | 5 (7,0) | 22 (8,1) | 16 (72,7) | 6 (27,3) |
| p.R1382Q (c.4145 G>A) | Sí | Sí | Presunto (causa clara) | 1 (1,4) | 2 (0,7) | 2 (100,0) | 0 |
| p.D928N (c.2782 G>A) | Sí | Sí | Presunto (causa clara) | 3 (4,2) | 13 (4,8) | 7 (53,8) | 6 (46,2) |
| p.E1348Q (c.4042 G>C) | Sí | No es posible | VUS2 (incierto) | 1 (1,4) | 4 (1,5) | 2 (50,0) | 2 (50,0) |
| p.E1356Q (c.4066 G>C) | Sí | No es posible | VUS2 (incierto) | 1 (1,4) | 4 (1,5) | 2 (50,0) | 2 (50,0) |
| p.E1387Q (c.4159 G>C) | Sí | No es posible | VUS2 (incierto) | 1 (1,4) | 4 (1,5) | 1 (25,0) | 3 (75,0) |
| p.R1079Q (c.3236 G>A) | Sí | No es posible | VUS2 (incierto) | 1 (1,4) | 2 (0,7) | 1 (50,0) | 1 (50,0) |
VSU: variante de significancia clínica desconocida (VUS2, incierto).
Los valores expresan n (%).
Se demostró un efecto fundador de las mutaciones c.2308+1G>A, p.P108Afs*9 y p.R891Afs*160 (c.2670-267insG) en MYBPC3 mediante análisis de haplotipo. El 74% de los portadores tenían en común 1 de estas 3 mutaciones fundadoras en la región, que causarían la formación de una proteína truncada. Se realizaron estudios de cosegregación y/o in silico en todas las variantes, con resultados compatibles con una mutación causante de enfermedad (tabla 1); 8 individuos (2,9%) presentaban una mutación adicional en otro gen.
La MCH se definió en los probandos como una HVI (≥ 15 mm) inexplicada, sin presencia de ninguna otra enfermedad cardiaca o sistémica que pudiera causar HVI1. Se consideró que los familiares estaban afectados si cumplían los criterios diagnósticos en el contexto de una MCH familiar24,25.
Se incluyó a los pacientes prospectivamente, y las exploraciones y los exámenes de diagnóstico por imagen cardiaca también fueron prospectivos; se registraron asimismo los síntomas y los eventos arrítmicos anteriores a la primera evaluación. La media de seguimiento fue de 5,5±3,3 años.
Tras el examen cardiaco, 192 individuos (el 70,6%; media de edad, 52,5±16,6 años; 127 [66,1%] varones) cumplían los criterios diagnósticos de MCH y se los consideró clínicamente afectados (pacientes), mientras que se consideró normales a 80 (29,4%), todos ellos portadores.
De los 260 individuos evaluados, 63 (23,2%) tenían HT. No se dispuso de información sobre la presión arterial de 12 de ellos (4,4%). Hubo 58 (31,7%) individuos hipertensos y 125 (68,3%) normotensos entre los 183 afectados de MCH con información disponible. De los 77 portadores normales con valores de presión arterial disponibles, 5 (3,3%) estaban hipertensos y 72 (90%), normotensos.
Actividad físicaSe preguntó a los participantes en el estudio acerca de su actividad física y se los clasificó según un concepto de «semana típica». Se tuvieron en cuenta únicamente las horas de actividad física que se estimó asociadas con un aumento > 70% de la frecuencia cardiaca máxima. Se tuvo en cuenta la actividad física durante los 2 años previos al momento del diagnóstico de los portadores afectados o hasta el momento de la primera evaluación de los portadores no afectados. Las entrevistas se realizaron prospectivamente en el momento de la primera evaluación de los pacientes visitados entre 2007 y 2015, y se contactó por teléfono con los pacientes evaluados entre 2003 y 2006. Se reclasificó como físicamente activos a los pacientes sedentarios en el momento de la evaluación pero con antecedentes de práctica deportiva de competición
- •
Grupo 1 (n=156 [84,9%]). Sedentarismo o actividad física baja (generalmente sin sudoración) (p. ej., caminar o cuidar el jardín): incluye a los participantes con un estilo de vida sedentario (n=137 [74,5%]) y aquellos con actividad física significativa ≤ 2 h/semana (n=19 [10,3%]).
- •
Grupo 2 (n=17 [9,3%]). Actividad física intermedia o moderada: incluye a los individuos con actividad física significativa > 2 y<5 h/semana (n=13 [7,1%]). Se incluyó en este grupo también a las personas que tenían un trabajo físicamente exigente (n=4 [2,2%]).
- •
Grupo 3 (n=11 [6%]). Actividad física intensa o alta: incluye a los deportistas aficionados, profesionales (n=6 [3,3%]) y exprofesionales (n=5 [2,7%]) que realizaban una actividad física regular intensa > 5 h/semana.
De los 71 portadores no afectados, 60 (84,5%) eran sedentarios o realizaban poca actividad física; 8 (11,3%) tenían actividad física intermedia o moderada y 3 (4,2%), intensa o alta. De 9 (11,2%) no se dispuso de información sobre la actividad física.
En la se muestra información detallada sobre el número de pacientes de cada subgrupo por lo que respecta a las variables de exposición (HT, sexo y actividad física), según la clasificación en «portador afectado»/«portador no afectado». Puede consultarse el .
Análisis estadísticoEl análisis estadístico se llevó a cabo con el programa estadístico SPSS (versión 15.0) y el programa STATA (versión 13) para el análisis de supervivencia de Laplace.
La edad al diagnóstico, la hipertrofia ventricular máxima y la MS se definieron como variables dependientes en el análisis. El sexo, la HT y la actividad física se definieron como variables exploratorias en el análisis multivariable. Los equivalentes de MS se definieron como MS, parada cardiaca reanimada o terapia apropiada de un desfibrilador automático implantable. La clase funcional de la New York Heart Association (NYHA) III-IV incluye los síntomas de insuficiencia cardiaca avanzada, trasplante de corazón y muerte relacionada con la insuficiencia cardiaca.
En el análisis multivariable de la supervivencia se incluyeron las 3 variables exploratorias y las seleccionadas como covariables. Para la penetrancia (edad al diagnóstico de MCH), se incluyó en el modelo el «tipo de gen» (MYBPC3 frente a MYH7) como variable independiente adicional. Para el ictus, la fibrilación auricular (FA), la «insuficiencia cardiaca (NYHA III-IV)», el «equivalente de MS» y los «eventos combinados» en los correspondientes análisis de supervivencia, las covariables consideradas incluyeron el grosor máximo de la pared indexado (mm/m2), el diámetro de la aurícula izquierda (AI) (mm), la presencia de obstrucción del tracto de salida del ventrículo izquierdo (TSVI) (> 30 mmHg) y el tipo de gen. Se utilizó un análisis de derivada de Laplace26 para el análisis de la supervivencia y la figura 2 y la figura 3. Se usó la mediana como parámetro de resumen en las comparaciones. En la evaluación de la influencia de las variables exploratorias en la HVI (grosor máximo de la pared en milímetros), se aplicó una regresión logística lineal. Se consideraron estadísticamente significativos los valores de p<0,05.
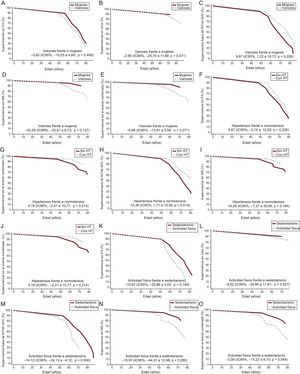
 en los portadores de mutaciones en MYBPC3 y MYH7. A: penetrancia según el sexo. B: presencia o ausencia de HT. C: intensidad de la actividad física. Se indica la mediana de Laplace ajustada y el IC95%. Las 3 variables exploratorias (sexo, HT y actividad física) y el «tipo de gen» se incluyeron en el análisis de cada uno de los gráficos. Se incluyó en el análisis a todos los portadores genéticos, afectados y no afectados. HT: hipertensión; IC95%: intervalo de confianza del 95%; MCH: miocardiopatía hipertrófica.")
Penetrancia de la enfermedad (MCH) en los portadores de mutaciones en MYBPC3 y MYH7. A: penetrancia según el sexo. B: presencia o ausencia de HT. C: intensidad de la actividad física. Se indica la mediana de Laplace ajustada y el IC95%. Las 3 variables exploratorias (sexo, HT y actividad física) y el «tipo de gen» se incluyeron en el análisis de cada uno de los gráficos. Se incluyó en el análisis a todos los portadores genéticos, afectados y no afectados. HT: hipertensión; IC95%: intervalo de confianza del 95%; MCH: miocardiopatía hipertrófica.
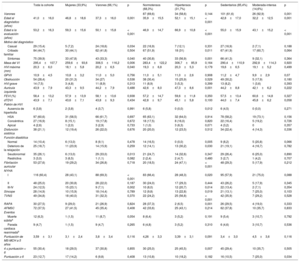
![Estimaciones de Laplace de la supervivencia ajustadas. A: sin FA. B: ictus. C: NYHA III-IV. D: MS. E: evento combinado según el sexo. F: sin FA. G: ictus. H: NYHA III-IV. I: MS. J: evento combinado según presencia o ausencia de HT. K: sin FA. L: ictus. M: NYHA III-IV. N: MS. O: evento combinado según la actividad física. Se indican los valores ajustados de la mediana de Laplace y el IC95%. Las 3 variables exploratorias (sexo, HT y actividad física) y las covariables (grosor máximo de la pared indexado [mm/m2], diámetro de la aurícula izquierda [mm], obstrucción del tracto de salida del ventrículo izquierdo [> 30 mmHg] y tipo de gen) se incluyeron en el análisis de cada uno de los gráficos, así como en los análisis. Solo se incluyó en el análisis a los portadores afectados. FA: fibrilación auricular; HT: hipertensión; IC95%: intervalo de confianza del 95%; MS: muerte súbita; NYHA: New York Heart AssociationI.](https://static.elsevier.es/multimedia/03008932/0000007100000003/v1_201803151140/S0300893217303603/v1_201803151140/es/main.assets/gr3.jpeg?xkr=eyJpdiI6Ik1UNU5RV0RtMFVoQlV6WmdHaFptaUE9PSIsInZhbHVlIjoiL2tPRjRGcDVQeE0wYXl4QWhhc3dPNTdwSkY2M1VYVkRWcVFKZmViRlkrR3RKUlNJQWRhbzhTaXp0N3NuVWZKNlhIbkFwd2FvU0R6NFVEUlFaZk83UTR1VUNmVDMzMXNVWVRVKzZhWEdFMkFEQWpvbGlDY21ZdEwyS2RYY05sNmxqWmZoaEhET083VjRGSXVnQmhMdXlCRURVekdEYUQxQzl3S3lJL215RHQ0WmpiSXQ2cWhySEJzeGt2QkZjQVlvamwwVncvZ1I3UU1vQ3JzNWxidkQrVk1iRUpkdHR2NGFRRXM2akJkRVRBbnNlK25ZZk5aeks2OU1kdHNrVUZ3Zi82bW56NDFNTVlHYUthbFN6Rk12R3JTWjJmZS9hZmlDa29MQnpLemc5UHc9IiwibWFjIjoiNjgxOWE0MjllMjIyYzI5ZTMxYjZjYzMwYzM4Y2YzZTRkNjQyZDg4MTRmMmI4ZDdiNjRhYjU3ZGI2NzJmY2ZhZSIsInRhZyI6IiJ9 "Estimaciones de Laplace de la supervivencia ajustadas. A: sin FA. B: ictus. C: NYHA III-IV. D: MS. E: evento combinado según el sexo. F: sin FA. G: ictus. H: NYHA III-IV. I: MS. J: evento combinado según presencia o ausencia de HT. K: sin FA. L: ictus. M: NYHA III-IV. N: MS. O: evento combinado según la actividad física. Se indican los valores ajustados de la mediana de Laplace y el IC95%. Las 3 variables exploratorias (sexo, HT y actividad física) y las covariables (grosor máximo de la pared indexado [mm/m2], diámetro de la aurícula izquierda [mm], obstrucción del tracto de salida del ventrículo izquierdo [> 30 mmHg] y tipo de gen) se incluyeron en el análisis de cada uno de los gráficos, así como en los análisis. Solo se incluyó en el análisis a los portadores afectados. FA: fibrilación auricular; HT: hipertensión; IC95%: intervalo de confianza del 95%; MS: muerte súbita; NYHA: New York Heart AssociationI.")
Estimaciones de Laplace de la supervivencia ajustadas. A: sin FA. B: ictus. C: NYHA III-IV. D: MS. E: evento combinado según el sexo. F: sin FA. G: ictus. H: NYHA III-IV. I: MS. J: evento combinado según presencia o ausencia de HT. K: sin FA. L: ictus. M: NYHA III-IV. N: MS. O: evento combinado según la actividad física. Se indican los valores ajustados de la mediana de Laplace y el IC95%. Las 3 variables exploratorias (sexo, HT y actividad física) y las covariables (grosor máximo de la pared indexado [mm/m2], diámetro de la aurícula izquierda [mm], obstrucción del tracto de salida del ventrículo izquierdo [> 30 mmHg] y tipo de gen) se incluyeron en el análisis de cada uno de los gráficos, así como en los análisis. Solo se incluyó en el análisis a los portadores afectados. FA: fibrilación auricular; HT: hipertensión; IC95%: intervalo de confianza del 95%; MS: muerte súbita; NYHA: New York Heart AssociationI.
Las características clínicas de los 192 pacientes en la primera evaluación por lo que respecta a las 3 variables de agrupación (sexo, HT y actividad física) se resumen en la tabla 227.
Principales características clínicas de los 192 pacientes con miocardiopatía hipertrófica evaluados
| Toda la cohorte | Mujeres (33,9%) | Varones (66,1%) | p | Normotensos (68,3%) | Hipertensos (31,7%) | p | Sedentarios (85,4%) | Moderada-intensa (14,6%) | p | |
|---|---|---|---|---|---|---|---|---|---|---|
| Varones | 87 (69,6) | 34 (58,6) | 0,144 | 101 (61,6) | 26 (92,9) | 0,001 | ||||
| Edad al diagnóstico (años) | 41,0±16,0 | 46,8±18,6 | 37,3±16,0 | 0,001 | 35,9±15,5 | 52,1±15,1 | < 0,001 | 42,8±17,0 | 32,2±12,5 | 0,001 |
| Edad a la evaluación (años) | 53,2±16,3 | 59,3±15,6 | 50,1±15,8 | < 0,001 | 46,9±14,7 | 66,9±10,8 | < 0,001 | 55,0±15,9 | 43,1±15,2 | < 0,001 |
| Motivo del diagnóstico | ||||||||||
| Casual | 29 (15,4) | 5 (7,2) | 24 (18,6) | 0,034 | 22 (16,9) | 7 (12,1) | 0,331 | 27 (16,9) | 2 (7,1) | 0,188 |
| Cribado familiar | 84 (44,7) | 30 (44,1) | 62 (41,8) | 0,504 | 67 (51,9) | 18 (31) | 0,011 | 67 (41,9) | 17 (60,7) | 0,064 |
| Síntomas | 75 (39,9) | 33 (47,8) | 43 (33,3) | 0,040 | 40 (30,8) | 33 (56,9) | 0,001 | 66 (41,3) | 9 (32,1) | 0,364 |
| Masa del VI | 295,4±107,7 | 259,6±93,6 | 306,5±116,2 | 0,006 | 283,4±122,2 | 306,7±85,9 | 0,164 | 290,4±110,9 | 292,8±114,3 | 0,920 |
| HVI máxima (mm) | 20,3±6,0 | 18,1±5,8 | 20,2±6,5 | 0,040 | 19,3±6,8 | 20,3±5,3 | 0,179 | 19,6±6,5 | 19,1±5,2 | 0,499 |
| GPVIi | 10,9±4,5 | 10,8±3,2 | 11,0±5,0 | 0,756 | 11,0±5,1 | 11,0±2,6 | 0,908 | 11,2±4,7 | 9,9±2,9 | 0,07 |
| Obstrucción | 54 (28,4) | 20 (31,3) | 34 (27) | 0,538 | 38 (30,4) | 15 (25,9) | 0,529 | 49 (30,2) | 5 (17,9) | 0,180 |
| HVI grave | 14 (7,5) | 3 (4,8) | 11 (8,9) | 0,313 | 11 (8,9) | 3 (5,3) | 0,399 | 13 (8,2) | 1 (3,6) | 0,393 |
| Aurícula izquierda | 43,9±7,9 | 43,3±9,5 | 44,2±7,9 | 0,488 | 42,6±8,0 | 47,3±8,6 | 0,001 | 44,2±8,8 | 42,1±6,2 | 0,222 |
| FEVI (%) | 58,4±13,2 | 57,9±13,9 | 58,1±13,6 | 0,938 | 57,2±14,7 | 59,6±11,8 | 0,350 | 57,5±13,4 | 60,6±14,8 | 0,327 |
| dTDVI | 43,9±7,1 | 43,6±7,1 | 43,8±9,3 | 0,434 | 42,8±9,7 | 45,1±5,8 | 0,100 | 44,0±7,4 | 45,8±6,2 | 0,058 |
| Patrón de HVI | ||||||||||
| Ausencia de hipertrofia | 6 (3,8) | 2 (3,8) | 4 (3,7) | 0,991 | 6 (5,8) | 0 (0,0) | 0,012 | 6 (4,5) | 0 (0,0) | 0,271 |
| HSA | 97 (60,6) | 31 (58,5) | 66 (61,7) | 0,697 | 65 (63,1) | 32 (64,0) | 0,914 | 78 (58,2) | 19 (73,1) | 0,156 |
| Concéntrica | 27 (16,9) | 8 (15,1) | 19 (17,8) | 0,672 | 18 (17,5) | 8 (16,0) | 0,820 | 22 (16,4) | 5 (19,2) | 0,726 |
| Apical | 4 (2,6) | 1 (2,0) | 3 (2,9) | 0,733 | 1 (1,0) | 3 (6,3) | 0,071 | 4 (3,2) | 0 | 0,357 |
| Disfunción sistólica | 38 (21,1) | 12 (19,4) | 26 (22,0) | 0,676 | 20 (20,0) | 12 (23,5) | 0,512 | 34 (22,4) | 4 (14,3) | 0,336 |
| Función diastólica | ||||||||||
| Normal | 14 (10,4) | 6 (13,0) | 8 (9,1) | 0,478 | 14 (16,5) | 0 (0,0) | 0,005 | 9 (8,2) | 5 (20,8) | 0,066 |
| Deterioro de la relajación | 25 (18,7) | 11 (23,9) | 14 (15,9) | 0,259 | 12 (14,1) | 13 (30,2) | 0,030 | 21 (19,1) | 4 (16,7) | 0,782 |
| Seudonormal | 35 (26,1) | 6 (13,0) | 29 (33,0) | 0,013 | 21 (24,7) | 14 (32,6) | 0,347 | 29 (26,4) | 6 (25,0) | 0,890 |
| Restrictiva | 3 (3,0) | 3 (6,5) | 1 (1,1) | 0,082 | 2 (2,4) | 2 (4,7) | 0,480 | 3 (2,7) | 1 (4,2) | 0,707 |
| Fibrilación auricular | 53 (27,6) | 19 (29,2) | 34 (26,8) | 0,718 | 20 (18,5) | 24 (47,1) | < 0,001 | 48 (29,3) | 5 (17,9) | 0,212 |
| NYHA | ||||||||||
| I | 116 (60,4) | 28 (43,1) | 88 (69,3) | < 0,001 | 83 (66,4) | 28 (48,3) | 0,020 | 95 (57,9) | 21 (75,0) | 0,088 |
| II | 48 (25,0) | 20 (30,8) | 28 (22,0) | 0,187 | 30 (24,0) | 17 (29,3) | 0,444 | 43 (26,2) | 5 (17,9) | 0,345 |
| III-IV | 24 (12,5) | 15 (23,1) | 9 (7,1) | 0,002 | 10 (8,0) | 12 (20,7) | 0,014 | 22 (13,4) | 2 (7,1) | 0,354 |
| Síncope | 28 (14,9) | 10 (15,9) | 18 (14,4) | 0,789 | 12 (9,8) | 13 (22,8) | 0,019 | 21 (13,1) | 7 (25,0) | 0,103 |
| TVNS | 49 (34,8) | 18 (40,0) | 31 (32,3) | 0,370 | 22 (24,2) | 25 (56,8) | < 0,001 | 42 (35,9) | 7 (29,2) | 0,528 |
| RAPA | 30 (27,5) | 9 (29,0) | 21 (26,9) | 0,824 | 28 (37,3) | 2 (6,5) | 0,001 | 26 (29,5) | 4 (19,0) | 0,333 |
| AFMSC | 72 (37,5) | 27 (41,5) | 45 (35,4) | 0,408 | 42 (33,6) | 25 (43,1) | 0,214 | 62 (37,8) | 10 (35,7) | 0,833 |
| Eventos | ||||||||||
| Muerte súbitaa | 12 (6,3) | 1 (1,5) | 11 (8,7) | 0,054 | 8 (6,4) | 3 (5,2) | 0,191 | 9 (5,4) | 3 (10,7) | 0,792 |
| Parada cardiaca reanimadab | 9 (4,7) | 1 (1,5) | 9 (4,7) | 0,265 | 6 (4,8) | 3 (5,2) | 0,310 | 6 (4,6) | 3 (10,7) | 0,536 |
| Puntuación de MS-MCH a 5 años | 3,59±3,1 | 3,1±2,4 | 3,8±3,4 | 0,116 | 4,26±3,3 | 3,39±3,1 | 0,091 | 3,4±3,0 | 4,5±3,6 | 0,116 |
| 4 ≤ puntuación < 6 | 55 (30,4) | 18 (29,5) | 37 (30,8) | 0,855 | 30 (25,0) | 25 (45,5) | 0,007 | 45 (29,4) | 10 (35,7) | 0,505 |
| Puntuación ≥ 6 | 23 (12,7) | 17 (14,2) | 6 (9,8) | 0,408 | 13 (10,8) | 10 (18,2) | 0,182 | 16 (10,5) | 7 (25,0) | 0,034 |
AFMSC: antecedentes familiares de muerte súbita cardiaca; DAI: desfibrilador automático implantable; dTDVI: diámetro telediastólico del VI; FEVI: fracción de eyección del VI; GPVIi: grosor de la pared del VI indexado; HSA: hipertrofia septal asimétrica; HVI: hipertrofia ventricular izquierda; MS-MCH: muerte súbita-miocardiopatía hipertrófica; NYHA: New York Heart Association; RAPA: respuesta anormal de la presión arterial; TVNS: taquicardia ventricular no sostenida; VI: ventrículo izquierdo.
Masa del VI según la fórmula de Deveraux (g). Obstrucción: si el gradiente del tracto de salida del VI > 30 mmHg. Patrón de HVI: subtipo morfológico de hipertrofia según Binder et al.27. HVI grave: si la HVI máxima es ≥ 30 mm.
Los valores expresan media±desviación estándar o n (%).
Las mujeres tenían una clase funcional de la NYHA superior tanto basal como en el seguimiento; 15 (23,1%) mujeres frente a 9 (7,1%) varones se encontraban en NYHA III-IV (p=0,002). El grosor máximo de la pared del VI indexado (GMPVI), el porcentaje de obstrucción, las variables de función sistólica y diastólica y la proporción de taquicardia ventricular no sostenida en la monitorización Holter fueron similares en las mujeres y los varones (tabla 2).
La proporción de individuos afectados aumentaba con la edad. A los varones se les diagnosticó la enfermedad 12,50 años antes que a las mujeres (IC95% de la mediana de Laplace, –17,52 a –6,48; p<0,001), ajustado por HT, actividad física y «tipo de gen» (figura 2A).
No hubo diferencias entre varones y mujeres por lo que respecta a la supervivencia sin FA, ictus, NYHA III-IV, MS y eventos combinados (FA, ictus, NYHA III-IV, MS) (figura 3A-3E).
HipertensiónDe los 183 pacientes información disponible sobre la HT que cumplían los criterios de MCH, 58 (31,7%) tenían HT (tabla 2). La presencia de síntomas fue la principal razón del diagnóstico de MCH de los hipertensos, en comparación con los normotensos (33 [56,9%] frente a 40 pacientes [30,8%]; p=0,001). Los pacientes con HT-MCH estaban en peor clase de la NYHA (12 pacientes hipertensos [20,7%] frente a 10 pacientes normotensos [8,0%] se encontraban en NYHA III-IV; p=0,01) y referían más síncopes (13 [22,8%] frente a 12 [9,8%]; p=0,014). No hubo diferencias en la HVI (masa del VI, GMPV o GMPVI) ni en la obstrucción del TSVI. Los pacientes con HT-MCH presentaban una HVI apical con una frecuencia ligeramente superior. Los diámetros telediastólicos de la AI y el VI estaban significativamente aumentados en los pacientes con HT-MCH en comparación con los normotensos (p=0,001 y p=0,082 respectivamente). Los pacientes con HT tenían más arritmias auriculares y ventriculares. Tenían FA 24 (47,1%) pacientes con HT-MCH, en comparación con 20 (18,5%) del grupo de normotensos (p<0,001). La taquicardia ventricular no sostenida en la monitorización Holter fue también más frecuente en los pacientes con HT-MCH (25 [56,8%] frente a 22 [24,2%]; p<0,001). Sin embargo, la respuesta anormal de la presión arterial fue más prevalente en los normotensos (28 [37,3%] frente a 2 [6,5%]; p=0,001).
El diagnóstico de la MCH fue más tardío en los pacientes hipertensos, con una diferencia de 10,83 años respecto a los normotensos (IC95% de la mediana de Laplace, 6,28-17,09; p<0,001), ajustada por sexo, actividad física y «tipo de gen» (figura 2B).
No hubo diferencias entre los pacientes con HT-MCH y los pacientes normotensos por lo que respecta a la supervivencia sin episodios de FA o ictus (figura 3F y figura 3G). Paradójicamente, la supervivencia sin NYHA III-IV fue menor en los pacientes normotensos que en aquellos con HT-MCH (p=0,02) (figura 3H), sin que hubiera diferencias significativas en la MS o el evento combinado (FA, ictus, NYHA III-IV, MS) (figura 3I y figura 3J).
Actividad físicaEn el momento de la evaluación, los pacientes con MCH que llevaban actividad física intensa tenían una edad significativamente inferior que los de los grupos más sedentarios (43,1±15,2 frente a 55,0±15,9 años; p=0,001). No hubo asociación alguna entre la intensidad del ejercicio y la HVI o la obstrucción del TSVI. Es de destacar que la dimensión de la AI y el diámetro telesistólico del VI fueron similares en los 2 grupos. Hubo una tendencia a una mejor función diastólica en el grupo de pacientes con MCH que realizaban una actividad física más intensa (flujo mitral normal, 5 [20,8%] frente a 9 [8,2%]; p=0,066).
No hubo diferencias significativas en los síntomas en relación con la intensidad de la actividad física. Las proporciones de pacientes con taquicardia ventricular no sostenida en la monitorización Holter y con FA fueron similares entre ambos grupos.
Aunque la proporción de individuos afectados fue similar, independientemente de la intensidad de la actividad física (el 72,2 frente al 71,8%), a los pacientes que realizaban una actividad física más intensa se les diagnosticó la MCH 7,34 años antes que a los del grupo sedentario (IC95% de la mediana de Laplace, –14,49 a–151; p=0,016), con ajuste por sexo, HT y «tipo de gen» (figura 2C).
No hubo diferencias entre estos 2 grupos por lo que respecta a la supervivencia sin episodios de FA, ictus, NYHA III-IV, MS o el evento combinado (FA, ictus, NYHA III-IV, MS) (figura 3K y figura 3O).
En la se presentan las características clínicas de 14 individuos que ejercicio intenso o de competición (11 afectados+3 portadores no afectados).
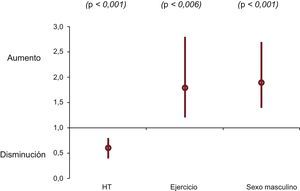
Análisis multivariableEn resumen, el sexo, la HT y la actividad física no mostraron valor predictivo respecto a la HVI en los análisis univariable y multivariable. Las 3 variables mostraban una asociación independiente con la edad al diagnóstico de la MCH en la regresión de Cox y en el análisis de supervivencia de Laplace (figura 4). Mientras que el sexo masculino y la actividad física intensa predijeron un diagnóstico más temprano, la HT se asoció con un retraso diagnóstico.
 obtenidos en el análisis multivariable de regresión de Cox incluyendo sexo, HT y actividad física. HT: hipertensión; IC95%: intervalo de confianza del 95%.")
Ninguna de las 3 variables exploratorias (sexo, HT, actividad física) se asoció con los eventos evaluados (ictus, FA, NYHA III-IV, MS o el evento combinado) en el análisis de regresión de Cox tras incluir como covariables el «tipo de gen», el diámetro de la AI, la HVI indexada y la obstrucción (). El diámetro de la AI fue la única variable independiente que mostró una asociación uniforme con los eventos evaluados. La HVI y el diámetro de la AI fueron factores independientes predictivos de MS en el análisis de regresión de Cox (HVI indexada, HR=1,10; IC95%, 1,00-1,20; p=0,039; diámetro de la AI, HR=1,06; IC95%, 1,03-1,10; p<0,001).
En el análisis multivariable de Laplace, el diámetro de la AI continuó siendo la principal covariable asociada con la supervivencia sin ictus, FA, NYHA III-IV y el evento combinado, pero no en relación con la MS (). El análisis de Laplace mostró también el valor de las 3 variables exploratorias respecto a la supervivencia NYHA III-IV. Con este análisis, ninguna de las variables exploratorias o las covariables mostró asociación con la supervivencia libre del evento combinado.
DISCUSIÓNLos mecanismos epigenéticos y los factores ambientales son fundamentales en las adaptaciones cardiacas y el desarrollo de la enfermedad cardiovascular28.
Este estudio presenta datos sobre la expresión de la enfermedad, la penetrancia (entendida como edad a la que se diagnostica la MCH) y la evolución clínica en una cohorte amplia de portadores de mutaciones asociadas a la MCH. Este es el primer y más amplio estudio que haya abordado la cuestión de la HT y el sexo en relación con la penetrancia y la gravedad del fenotipo, y es el único que ha investigado la influencia de la actividad física intensa en la edad al diagnóstico, la gravedad de la enfermedad y la evolución clínica en una serie de pacientes con MCH genotipificada. En este estudio se incluyó a 272 pacientes genotipificados con 3 mutaciones fundadoras con una repercusión muy similar en la proteína (MYBPC3 IVS23+1G>A, p.R891Afs*160, p.P108Afs*9), que suponen un 74% de la población (n=199 [85+65+49]).
SexoEn consonancia con lo indicado por estudios previos17-21,29-32, se observa que el sexo masculino se asocia con un diagnóstico de MCH más temprano. En esta serie, se diagnosticó a los varones más de una década antes que a las mujeres. Las posibles explicaciones de esta observación uniforme podrían estar en el papel de los estrógenos en el desarrollo de la hipertrofia33.
Una vez desarrollado el fenotipo, las mujeres evolucionan de manera similar que los varones, alcanzan un similar grado de HVI (indexada) y obstrucción y muestran una limitación sintomática aún mayor. No se identificaron diferencias constantes entre los sexos en cuanto a insuficiencia cardiaca, FA e ictus. Al igual que en la literatura médica previa, en nuestra serie no se evidenciaron diferencias significativas por sexo en la MS y el evento combinado.
HipertensiónExiste la creencia general, basada en datos indirectos o en series pequeñas, de que la HT debería asociarse con un aumento de la HVI en los pacientes con MCH34–36 y que la actividad física intensa se asocia con un aumento de la HVI y es también un desencadenante de arritmias malignas y, por consiguiente, una causa de MS en la MCH37,38.
Un artículo recientemente publicado sobre el papel de la HT y el sexo del paciente en la penetrancia y el fenotipo ha mostrado asociación bivariable entre la HT y la expresión de la MCH35. El estudio no observó diferencias entre varones y mujeres ni según la actividad física. No fue posible realizar un análisis multivariable debido al pequeño tamaño muestral, y hubo un porcentaje significativo de datos no disponibles (44%). En otro artículo reciente sobre el tema36, no se realizó un análisis estadístico. En esta segunda serie tampoco se registró la actividad física.
Hasta donde se sabe, este es el primer estudio que demuestra claramente el efecto de otros factores de riesgo adicionales en los resultados fenotípicos de una población con MCH genotipificada y homogénea. Para los individuos hipertensos de nuestra serie, el diagnóstico de MCH fue más tardío. Esta observación no era totalmente inesperada, ya que la hipertrofia podría considerarse una consecuencia de la HT en algunos pacientes hipertensos con fenotipos leves. Además, la medicación administrada a los pacientes con HT podría prevenir o retrasar la manifestación del fenotipo de MCH. El diseño del estudio impide extraer otras conclusiones. Aunque para el análisis se dispuso de la edad al diagnóstico, no hubo datos sobre la edad a la aparición de la HVI.
Aun cuando los pacientes con HT-MCH presentaban más síntomas y tenían mayor prevalencia de arritmias, en el análisis de supervivencia no parece que eso influyera en el pronóstico, en comparación con los pacientes normotensos tras ajustar por las covariables.
Actividad físicaEl diagnóstico de MCH se incluye en las guías actuales como causa de exclusión de deportes de competición39–41. Esta recomendación, que se amplía a los portadores asintomáticos, se basa en la hipótesis de que un entrenamiento de ejercicio regular y la participación en deportes de competición pueden desempeñar un papel en el desencadenamiento de mecanismos celulares que conduzcan al fenotipo de MCH y a eventos arrítmicos en presencia de una anomalía génica predisponente. Los autores de estas guías concluyen que no hay datos sólidos que respalden esta recomendación y que esta se basa en lo que parece más razonable a los grupos de expertos42.
En la presente serie, se observa que a los portadores de mutaciones sarcoméricas de los grupos de ejercicio más intenso se les diagnosticó MCH 7 años antes que a los portadores sedentarios.
Una vez aparecida la enfermedad, en los pacientes con MCH que realizan actividad física en los grupos de mayor intensidad se desarrolla similar grado de hipertrofia. Tiene interés señalar que, en el análisis multivariable, el sexo, la HT y la intensidad de la actividad física no tuvieron valor predictivo de HVI en esta serie. Es interesante que no se identificaran diferencias en las dimensiones del VI y la AI entre los grupos de MCH con actividad física y sedentario. La miocardiopatía hipertrófica podría impedir el remodelado cardiaco de los deportistas. La actividad física parece tener una repercusión mayor en el tamaño del ventrículo derecho de los individuos normales y los portadores de mutaciones desmosómicas, en comparación con la que tiene en el VI de los pacientes con MCH42. En cambio, las 3 variables mostraron una asociación significativa con la edad al diagnóstico de MCH.
Hay poca información en la literatura médica sobre el presunto mal pronóstico de los pacientes con MCH que realizan una actividad física intensa, y la que hay procede de informes clínicos y series retrospectivas43.
En el presente estudio, los pacientes con MCH que realizaban una actividad física más intensa mostraron similar frecuencia de eventos (FA, ictus, NYHA III-IV, MS y eventos combinados).
En conclusión, según lo indicado por los datos obtenidos, la actividad física sí desempeña un papel en lo relativo a la edad al diagnóstico, pero no es significativa en cuanto a la expresión de la enfermedad y, en particular, no parece que tenga una repercusión importante en el pronóstico. La explicación de esta observación deberá buscarse en futuros estudios prospectivos más amplios.
LimitacionesNo fue posible evaluar el efecto de las dobles mutaciones, ya que la mayor parte de los estudios genéticos se realizaron en la época previa a la ultrasecuenciación. Se realizó un análisis de 2 a 5 genes (incluidos en todos los casos MYH7 y MYBPC3) en el 80% de los pacientes, mientras que en un 20% de los casos índice, el estudio genético se consideró completo cuando se identificaba 1 de las mutaciones fundadoras. No fue posible evaluar la edad a la aparición de la enfermedad, ya que el periodo de estudio era limitado. Se utilizó la edad al diagnóstico como indicador sustituto de la penetrancia de la enfermedad. Las definiciones de la intensidad de la actividad física fueron arbitrarias. La clasificación tuvo como objetivo evaluar la repercusión de los hábitos de ejercicio en el momento del diagnóstico y la gravedad de la enfermedad en los resultados clínicos. El análisis de la relación dosis-efecto por lo que respecta al deporte o la actividad física quedó fuera del ámbito de este artículo. El número de individuos en el grupo de actividad física intensa fue bajo.
CONCLUSIONESA los varones y los portadores de mutaciones de MCH que realizan una actividad física intensa se los diagnostica significativamente antes que a las mujeres y los familiares sedentarios. En cambio, se diagnostica más tarde a los hipertensos portadores de mutaciones de MCH. El sexo, la HT y el grado de actividad física no muestran una asociación significativa con la gravedad de la HVI en los portadores de mutaciones causales de MCH.
FINANCIACIÓNEste estudio fue financiado por una subvención de la Fundación Española del Corazón-Coca-Cola LTD (2007) y contó con el apoyo de la Fundación para la Formación e Investigación Sanitaria de la Región de Murcia. Los investigadores forman parte de la red de investigación cardiovascular del Instituto de Salud Carlos III (RIC; RD12/0042/0049) y el IMIB (Instituto Médico de Investigación Biosanitaria). M. Sabater recibió una subvención de ISCIII (RIC). Los investigadores forman parte del grupo clínico de CIBERER y la Universidad de Murcia.
CONFLICTO DE INTERESESNo se declara ninguno.
- –
Los varones portadores de mutaciones de MCH contraen la enfermedad en una fase de la vida más temprana que las mujeres portadoras. Existe la creencia general de que la HT y el ejercicio intenso se asocian con un aumento de la HVI en los pacientes con MCH. Hay datos indirectos que respaldan la idea de que una actividad física intensa podría desencadenar arritmias ventriculares causantes de MS en los pacientes con MCH.
- –
Este estudio pone de manifiesto que, además del sexo, la HT y la actividad física también pueden tener influencia en la edad al diagnóstico de MCH de los portadores genéticos. No parece que sea significativo el papel de la HT y la actividad física en relación con la gravedad del fenotipo y la evolución clínica.
Los autores desean expresar su agradecimiento a Iván Gómez, M. José Oliva, María López, M. Carmen Olmo, M. José Antolinos y Miguel Pagan por su ayuda en el trabajo clínico y molecular de este proyecto. Damos las gracias en especial a los pacientes y al Biobanco (PT13/0010/0018) de la Red Nacional de Biobancos de España (B.000859).