
El síndrome de QT largo es una canalopatía hereditaria que se asocia a síncope y muerte súbita. La heterogeneidad fenotípica de esta enfermedad hace que el estudio genético sea fundamental para detectar a los sujetos con síndrome de QT largo oculto. En este trabajo se exponen las características de una familia con 13 portadores de la mutación missense KCNH2-H562R que afecta a la región del poro del canal HERG.
MétodosSe describió la mutación KCNH2-H562R en un varón de 65 años con intervalo QTc prolongado que presentó un episodio de torsade de pointes. Posteriormente, se identificaron 13 portadores de la mutación en la familia. Se realizó evaluación clínica, electrocardiograma y ecocardiograma a los portadores (edad, 48±26 años; el 46% varones).
ResultadosEl QTc medio en los portadores fue de 493±42ms (3 [23%] mostraron QTc normal); 6 (46%) tuvieron síntomas (4, síncope; 1, muerte súbita; 1, muerte súbita resucitada [probando]). Durante el tratamiento con bloqueadores beta, 11 (92%) de los 12 portadores permanecieron asintomáticos a los 5 años de seguimiento (1 paciente requirió simpatectomía cardiaca izquierda). El acortamiento del QTc con bloqueadores beta fue de 50±37ms. Hubo 1 muerte súbita en un paciente que rechazó tratamiento con bloqueadores beta.
ConclusionesEl estudio familiar es fundamental en la interpretación de los resultados de los tests genéticos en la actualidad. Este artículo describe el fenotipo variable y heterogéneo de una amplia familia portadora de la mutación KCNH2-H562R y destaca el papel del estudio genético en la identificación de los individuos en riesgo que se beneficiarían del tratamiento con bloqueadores beta.
Palabras clave
El síndrome de QT largo (SQTL) congénito es una canalopatía hereditaria que se caracteriza por una prolongación de la repolarización ventricular, que se manifiesta por un incremento del intervalo QT en el electrocardiograma (ECG). Esto expone a los pacientes a presentar síncope y muerte súbita por torsade de pointes que degenere en fibrilación ventricular. Hasta la fecha, se han identificado más de 700 mutaciones en 13 genes relacionados con SQTL; los genotipos más frecuentes son los de SQTL tipos 1–31. El SQTL tipo 2 (QTL2) se asocia a mutaciones de pérdida de función del gen human ether-a-go-go related (HERG; KCNH2; Kv11.1)2 y se presenta en un 30–45% del total de pacientes con genotipo positivo para SQTL1. El gen HERG codifica la subunidad α del canal de potasio dependiente del voltaje de la corriente de K+ rectificadora tardía cardiaca. Cada subunidad α está formada por seis segmentos transmembrana alfa-helicoidales (S1-S6), en los que el poro del canal está entre los segmentos S5 y S62. Se ha demostrado que las mutaciones del gen KCNH2 que afectan a la región de poro-bucle del canal, encargada de la vía de conducción de iones, se asocian a un aumento del riesgo de eventos arrítmicos3,4. Además, la evolución natural de la enfermedad difiere en función del sexo y la edad5 y con frecuencia hay una expresión génica variable en los familiares que comparten la misma mutación. Por otra parte, la información existente sobre la correlación entre genotipo y fenotipo suele basarse en casos aislados y en árboles genealógicos pequeños. El objetivo de este estudio es describir los eventos cardiacos, sus desencadenantes y las características del ECG de una amplia familia con QTL2 y, en segundo lugar, evaluar la respuesta al tratamiento con bloqueadores beta.
MÉTODOSPoblación en estudioEl probando era un varón de 65 años que consultó por un episodio de taquicardia ventricular polimórfica después de 3 días en tratamiento con un macrólido. El ECG tras la taquicardia mostró un intervalo QTc de 600ms. Se elaboró el árbol genealógico del paciente y se ofreció a los familiares un examen o cribado de detección de SQTL. La evaluación del probando y sus familiares incluyó exploración física, ECG de 12 derivaciones, cálculo de la puntuación de Schwartz6 y ecocardiografía Doppler bidimensional. Se obtuvieron muestras de sangre para el análisis genético y se obtuvo el consentimiento informado del paciente y sus familiares. El estudio fue aprobado por el comité ético local.
Se determinó el intervalo QT en las derivaciones II y V5 o V6, y se corrigió por la frecuencia cardiaca aplicando la fórmula de Bazett (QTc = QT/RR0,5). Se consideró intervalo QTc largo todo valor>450ms en varones y > 460 en mujeres7. Según el último documento de consenso europeo de síndromes arrítmicos hereditarios primarios8, el diagnóstico de SQTL en el probando se basó en la presencia de una puntuación de riesgo de SQTL ≥ 3,56 y un intervalo QT corregido ≥ 500ms en ECG repetidos. Tras la identificación de la mutación KCNH2-H562R en el probando, el diagnóstico en los familiares se realizó mediante el estudio genético de la mutación.
Estudio genéticoSe extrajo ADN genómico de muestras de sangre periférica mediante protocolos estándares. Se amplificaron todos los exones de los genes KCNQ1, KCNH2 y SCN5A con cebadores intrónicos y se secuenciaron en ambas direcciones empleando el kit BigDye v1.1 y el analizador ABI3130 (Applied Biosystems; Foster City, California, Estados Unidos). Se comparó la secuencia de cada gen con la secuencia de referencia de la base de datos del National Center for Biotechnology Information. Se evaluó la patogenicidad de la nueva variante mediante estudio in silico.
Análisis estadísticoSe utilizó el programa estadístico SPSS versión 15.0 para el análisis estadístico de los resultados (SPSS; Chicago, Illinois, Estados Unidos). Se evaluó con la prueba de Kolmogorov-Smirnov si las variables continuas tenían distribución normal. Las variables con distribución normal se expresan como media±desviación estándar y las variables cualitativas, como valores absolutos y porcentajes. Se utilizó la prueba de la U de Mann-Whitney para objetivar la presencia de diferencias entre los portadores y los no portadores de la mutación KCNH2-H562R. El acortamiento del QTc en tratamiento con bloqueadores beta se evaluó mediante el test de Wilcoxon y el efecto de los diferentes tipos de bloqueadores beta, con la prueba de Kruskal-Wallis. Se consideraron estadísticamente significativos los valores de p<0,05.
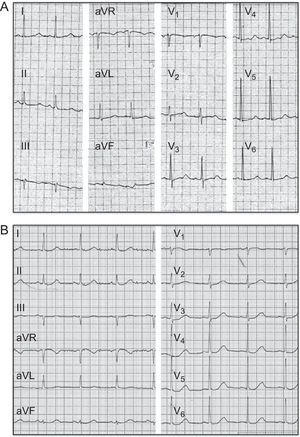
RESULTADOSHistoria clínicaEl paciente índice (II.2) era un varón caucásico de 65 años de edad que ingresó en el hospital por un episodio sincopal mientras caminaba. Se había tratado al paciente con claritromicina durante los 3 días previos debido a una infección respiratoria. Refirió haber presentado varios episodios de síncope desde los 30 años de edad, pero nunca lo habían estudiado por ello. El análisis de sangre reveló hipopotasemia leve (K+, 3,1 mEq/l), sin otras alteraciones. Mientras se encontraba en el área de observación del servicio de urgencias, el paciente presentó un episodio de taquicardia ventricular polimórfica que degeneró en fibrilación ventricular y requirió cardioversión eléctrica. En el ECG obtenido tras la cardioversión, se observó un intervalo QTc de 600ms (figura 1A). Posteriormente se suspendió el tratamiento con claritromicina y se corrigió la hipopotasemia, puesto que ambos factores producen una prolongación anormal del intervalo QT. Sin embargo, el intervalo QTc continuaba estando claramente prolongado (520ms), por lo que se inició tratamiento con bisoprolol.

En tratamiento con dosis media de bisoprolol, el QTc se redujo a 490ms (figura 1B). Sin embargo, el paciente presentaba bradicardia sinusal sintomática y múltiples episodios de presíncope ortostático. Por consiguiente, se decidió implantar un marcapasos DDD-R (el paciente rechazó implante de un desfibrilador automático implantable) para aumentar la frecuencia cardiaca y la dosis de bloqueadores beta, y así conseguir una mayor reducción del intervalo QTc y controlar los síntomas secundarios a la bradicardia sinusal. Con estimulación auricular y dosis máxima de bisoprolol, el QT se redujo a 470ms. Después el paciente ha permanecido asintomático, no ha presentado ningún evento arrítmico en los ECG Holter de 24h ni en las pruebas de esfuerzo realizados anualmente en un periodo de seguimiento de 5 años.
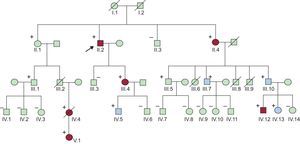
Se construyó el árbol genealógico del paciente y se realizó el cribado de SQTL en los familiares de primer grado (figura 2). En el probando se llevó a cabo un análisis genético de los principales genes de canales iónicos cardiacos causantes de SQTL. Dicho análisis reveló que el caso índice era heterocigoto para la mutación missense KCNH2-H562R, por lo que se diagnosticó de QTL2.
. En azul, los portadores asintomáticos con intervalo QTc prolongado, y en rojo, los pacientes que presentaron síntomas (síncope, muerte súbita, muerte súbita recuperada). Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Árbol genealógico. Los pacientes III.6, III.8 e III.9 fallecieron antes de cumplir 1 año de edad. Los portadores de la mutación aparecen marcados con (+). En azul, los portadores asintomáticos con intervalo QTc prolongado, y en rojo, los pacientes que presentaron síntomas (síncope, muerte súbita, muerte súbita recuperada). Esta figura se muestra a todo color solo en la versión electrónica del artículo.
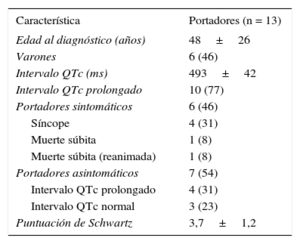
Se realizó el cribado en 24 familiares (figura 2) de la mutación previamente identificada en el probando, y 12 de ellos (50%) eran portadores de la mutación KCNH2-H562R. Las características demográficas, los intervalos QTc y los eventos clínicos en los portadores de la mutación se muestran en la tabla 1.
Características clínicas y electrocardiográficas de los portadores de la mutación KCNH2-H562R
| Característica | Portadores (n = 13) |
|---|---|
| Edad al diagnóstico (años) | 48±26 |
| Varones | 6 (46) |
| Intervalo QTc (ms) | 493±42 |
| Intervalo QTc prolongado | 10 (77) |
| Portadores sintomáticos | 6 (46) |
| Síncope | 4 (31) |
| Muerte súbita | 1 (8) |
| Muerte súbita (reanimada) | 1 (8) |
| Portadores asintomáticos | 7 (54) |
| Intervalo QTc prolongado | 4 (31) |
| Intervalo QTc normal | 3 (23) |
| Puntuación de Schwartz | 3,7±1,2 |
Los valores expresan media±desviación estándar o n (%).
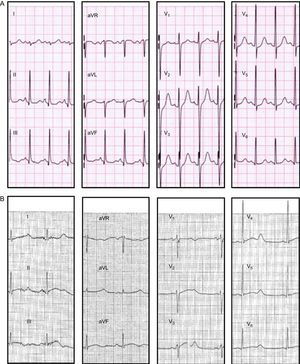
En el estudio familiar se descubrieron tres muertes inexplicadas en niños menores de 1 año (III.6, III.8, III.9) (el estudio genético no fue posible y no se dispuso de los ECG). De los 13 portadores de la mutación, 6 (el 46%, incluido el caso índice) tenían síntomas cardiacos (tabla 2). La paciente IV.4 se había diagnosticado de epilepsia por varios episodios nocturnos de convulsiones. Esta paciente mostraba una amplia variación del intervalo QTc en ECG previos (QTc entre 430 y 480ms), por lo que se le prescribió tratamiento con bloqueadores beta, que rechazó. Una noche mientras dormía, sonó el teléfono, y la paciente perdió el conocimiento poco después de iniciar la conversación; 6 años después, la paciente falleció súbitamente mientras dormía. Su padre (III.2), que debía ser portador obligado de la mutación, había fallecido súbitamente a los 35 años de edad en su casa 15 años antes (no se realizó autopsia). La hija de la paciente IV.4 (paciente V.1) había presentado varios síncopes en reposo a pesar del tratamiento con propanolol (QTc, 480ms) (figura 3A) y requirió denervación simpática cardiaca izquierda a la edad de 6 años, puesto que se la consideró demasiado joven para el implante de un desfibrilador automático implantable.
Eventos en familiares portadores de la mutación KCNH2-H562R
| Paciente | Edad en la primera valoración (años) | QTc en la primera valoración (ms) | Síntomas |
|---|---|---|---|
| II.4 | 83 | 508 | Síncope en reposo |
| III.4 | 53 | 490 | Síncope en reposo |
| IV.4 | 23 | 455 | Episodios convulsivos (diagnosticada de epilepsia); síncope desencadenado por un estímulo acústico; muerte súbita |
| IV.12 | 20 | 600 | Síncope en reposo |
| V.1 | 3 | 488 | Síncope en reposo |
. B: paciente IV.12 (QTc, 600ms). Ambos pacientes presentaron síncopes en reposo.")
Actualmente, todos los portadores vivos (n = 12) están en tratamiento con bloqueadores beta y solo la paciente V.1ha tenido síntomas cardiacos durante un seguimiento de 5 años.
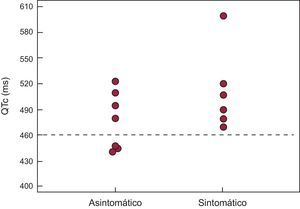
Estudio electrocardiográfico de los portadores de la mutaciónSe observaron muescas típicas de SQTL en la onda T en el ECG de 3 (23%) pacientes (figura 3B), y las ondas T anchas eran la morfología predominante en los demás. El intervalo QTc fue mayor en los portadores de la mutación que en los familiares no portadores (QTc, 493±42 frente a 418±14ms; p<0,001). El QTc fue más prolongado en los portadores sintomáticos que en los asintomáticos, si bien la diferencia no alcanzó la significación estadística (QTc, 517±57 frente a 477±33ms; p = 0,28) (figura 4). El intervalo QTc fue normal en el 23% de los portadores (n = 3), todos ellos asintomáticos. Las mujeres mostraron un QTc más largo que los varones, pero esta diferencia tampoco alcanzó la significación estadística (QTc, 508±57 frente a 481±21ms; p = 0,23).

En espera de los resultados del estudio genético, se realizó test de esfuerzo a los familiares con QTc normal, con objeto de identificar a posibles portadores. En los 3 individuos con QTc normal que posteriormente se mostraron portadores de la mutación, el intervalo QTc mostró un comportamiento similar con el ejercicio: durante el primer minuto el QTc aumentó 25±5ms, luego se redujo progresivamente durante el resto del periodo de ejercicio, y en el tercer minuto de la fase de recuperación aumentó 25±10ms. Este comportamiento patológico permitió el inicio precoz de tratamiento con bloqueadores beta en estos pacientes.
Todos los portadores recibieron bloqueadores beta (excepto la paciente IV.4, como ya se ha señalado). De ellos, en 5 (42%) se inició tratamiento con bisoprolol; en 4 (33%), con metoprolol, y en 3 (25%), con propranolol (se excluyó del análisis a 1 paciente por presentar estimulación ventricular por marcapasos). El acortamiento medio del QTc con el uso de bloqueadores beta fue de 50±37ms (n = 11; QTc, 493±46 frente a 442±19ms; p = 0,002). Dicho acortamiento fue de 44±25ms con bisoprolol, 47±23ms con metoprolol y 63±73ms con propranolol (p = 0,9).
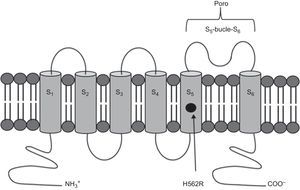
Análisis genéticoSe identificó la variante heterocigota c.1685A > G, p.H562R (rs199472922) en el exón 7 del gen KCNH2. Esta variante causa un cambio en el aminoácido 562 de histidina a arginina, sustitución que afecta a la región transmembrana S5 de la proteína (figura 5). El aminoácido involucrado se encuentra en una región altamente conservada entre especies y genes parálogos. El estudio bioinformático de la mutación (Mutation Taster), mostró que se trataba de una variante patogénica con p = 0,99 (p es el valor de predicción; los valores próximos a 1 indican alto grado de fiabilidad de la predicción realizada).
DISCUSIÓN
En este estudio se describe por primera vez el fenotipo de la mutación missense KCNH2-H562R en una amplia familia con SQTL. Así pues, a partir de los datos clínicos de un probando con antecedentes de síncope que experimenta una muerte súbita reanimada en el contexto de hipopotasemia y tratamiento con claritromicina, se realiza un estudio genético y familiar exhaustivo.
Implicaciones de las mutaciones del HERG localizadas en la región de poroEn humanos, el gen KCNH2 está situado en el cromosoma 7q35-36 y tiene una región codificante de 16 exones2. La subunidad HERG1 está compuesta por 1.159 aminoácidos y contiene seis dominios transmembrana (S1-S6). El poro selectivo de K+ se encuentra entre los dominios transmembrana S5 y S6 (aminoácidos 550 a 650). La mutación identificada en la familia estudiada está situada en el dominio S5 y forma parte del poro del canal. En 2002, Moss et al3 publicaron el seguimiento a 40 años de 44 mutaciones diferentes en HERG, y observaron que los individuos con mutaciones en el poro presentaban manifestaciones clínicas más graves y mayor frecuencia de episodios arrítmicos (el 74 frente al 35%; p<0,001). Posteriormente, en un estudio en el que se incluyó a 858 pacientes con QTL2 y 162 mutaciones diferentes del gen KCNH2, Shimizu et al4 describieron que los pacientes de más riesgo eran aquellos con mutaciones en la región S5-bucle-S6. El principal motivo es que las mutaciones que afectan al poro del canal tienen mayor efecto negativo en la corriente rectificadora tardía de K+. Sin embargo, solo un pequeño porcentaje de las mutaciones del gen KCNH2 se han caracterizado mediante estudio electrofisiológico funcional in vitro2,4.
La mutación KCNH2-H562R se describió en 2 pacientes aislados en un estudio en el que se analizó el espectro de mutaciones halladas en 2.500 casos de SQTL no relacionados9, aunque en ese registro no se detalla la correlación fenotipo-genotipo de las variantes genéticas halladas. Por otro lado, Sharma et al10 realizaron el estudio funcional de otra mutación en el mismo aminoácido (H562P) en un paciente con varios episodios de síncope, y observaron una marcada reducción de la corriente de K+. Todos estos hallazgos respaldan la patogenicidad de la mutación hallada en la familia que se describe en este artículo.
La evaluación de la patogenicidad de una variante genética identificada a veces es controvertida, y más ahora tras la introducción de las nuevas técnicas de secuenciación de ADN (Next Generation Sequencing)11,12. Con estas técnicas de secuenciación, se puede analizar un amplio número de genes; con lo que podrían aumentar los problemas de interpretación. Así, estudios recientes han puesto en duda la patogenicidad de hasta un 17% de las variantes genéticas catalogadas previamente como causales de miocardiopatías11,12. Este artículo muestra cómo la información clínica y el análisis de cosegregación de la mutación con la enfermedad permitieron establecer el diagnóstico.
Correlación genotipo-fenotipoEl estudio del ECG mostró una frecuencia de ondas T melladas menor que lo descrito en la bibliografía13: solo un 23% de los portadores presentaban dicho patrón. Además, se observó una gran variación en el fenotipo y la duración del intervalo QTc en los familiares portadores de la misma mutación. Esta penetrancia variable podría explicarse en parte por la coexistencia de polimorfismos de nucleótido único en los genes causantes del QTL o genes no conocidos o mutaciones múltiples14.
No han observado diferencias en la dución del QTc entre los portadores con y sin síntomas, de tal manera que con el valor del intervalo QTc no se podría identificar a priori a los individuos que tendrían síntomas en el seguimiento. Además, 3 portadores asintomáticos mostraron un QTc normal. En este contexto, se ha descrito que existe un solapamiento importante en la distribución del QTc entre individuos sanos y pacientes con un SQTL confirmado genéticamente. Así pues, según estudios previos, un 40% de los pacientes con SQTL y genética identificada muestran valores de QTc<460ms15.
Múltiples estudios han demostrado la utilidad de la respuesta del QTc al ejercicio en los pacientes con SQTL. Los pacientes con SQTL tipo 1 frecuentemente muestran una marcada prolongación del QTc durante el ejercicio, mientras que en los pacientes con QTL2 este incremento es discreto o inexistente16. Posteriormente se describió que el intervalo QT se prolonga a los 2–3min de la fase de recuperación en los pacientes con QTL217,18, comportamiento también objetivado en los portadores de la familia descrita en este artículo.
El sexo se ha identificado como factor independiente importante en el desarrollo de eventos en el QTL2, de tal manera que las mujeres presentan un riesgo de eventos cardiacos significativamente superior al de los varones tras el inicio de la adolescencia5. Esta observación se corrobora en este estudio, en el que las mujeres presentaron un QTc más prolongado y 4 de los 6 portadores sintomáticos (66%) eran mujeres.
La mayor parte de los eventos cardiacos en los pacientes con QTL2 se asocian a situaciones de aumento del tono simpático, estrés emocional o estímulos auditivos súbitos19, como en el caso de la paciente IV.4. Un estudio publicado en 201020 puso de manifiesto que los más importantes factores de riesgo de presentar eventos cardiacos desencadenados con el despertar son el sexo femenino y las mutaciones del poro-bucle, mientras que para los eventos desencadenados con el ejercicio, los más importantes son la presencia de las mutaciones que no afectan al poro-bucle del canal. Sin embargo, otros portadores de la mutación estudiada presentaron síntomas en reposo. En este contexto, varios estudios han descrito que alrededor de un 30% de los eventos cardiacos de los pacientes con un QTL2 se producen en reposo21. Así, el sueño REM se asocia a una activación simpática profunda, lo cual podría desempeñar un papel importante en el desencadenamiento de eventos cardiacos en los pacientes con QTL2.
EpilepsiaNo es infrecuente que se diagnostique epilepsia a los pacientes con SQTL. En este sentido, el gen KCNH2 se descubrió inicialmente en el hipocampo, y su expresión se demostró después en muchas regiones del sistema nervioso central. En un estudio en el que se incluyó a 343 probandos con SQTL, los antecedentes personales de crisis epilépticas fueron más frecuentes en el QTL2 que en todos los demás subtipos de SQTL juntos22. En la familia estudiada, la paciente IV.4 tenía antecedentes de epilepsia tratada farmacológicamente. Algunos fármacos antiepilépticos, entre los que destaca la fenitoína, tienen potencial arritmogénico debido a que son potenciales bloqueadores de la corriente de K+ rectificadora tardía cardiaca in vitro. En consecuencia, debe realizarse un ECG a todos los pacientes con síncope y crisis epilépticas con el objetivo de descartar un SQTL2, puesto que el tratamiento con fármacos antiepilépticos puede ser inadecuado y perjudicial.
Otros desencadenantesExisten múltiples causas de prolongación anormal del intervalo QT, como la isquemia miocárdica, las miocardiopatías, la hipotermia, la hipopotasemia, la hipocalcemia, la hipomagnesemia, el sistema nervioso autónomo, algunos fármacos y la recientemente descrita disminución de la temperatura epicárdica23. La hipopotasemia y la claritromicina fueron posiblemente los factores desencadenantes del evento arrítmico del paciente índice. Algunos estudios han descrito recientemente una reducción del intervalo QTc en los pacientes con QTL2 con una combinación de suplementos de potasio e inhibidores de la aldosterona. En la familia descrita se recomendó el tratiento con suplementos de potasio al probando y otros portadores con hipopotasemia o concentraciones de potasio próximas al límite inferior de la normalidad. Por otro lado, existe una amplia gama de fármacos que prolongan el intervalo QT y/o inducen taquicardia ventricular polimórfica24. En este contexto, se ha descrito un intervalo QT largo y arritmias ventriculares en pacientes tratados con eritromicina por una inhibición del canal de la corriente rectificadora tardía de K+25. De manera similar a lo observado en el paciente índice, se han publicado varios casos de prolongación del intervalo QT y torsade de pointes tras la administración de claritromicina. Dado que la estructura de la claritromicina es similar a la de la eritromicina, las propiedades arritmogénicas deben ser similares en ambos fármacos26. Otros macrólidos como la roxitromicina y la azitromicina parecen ser menos arritmogénicos en los estudios comparativos realizados in vitro27.
TratamientoEl tratamiento del SQTL tiene como objetivo reducir la incidencia del síncope y muerte súbita. El tratamiento estándar incluye el uso de bloqueadores beta, en ocasiones asociado al implante de un desfibrilador automático implantable28. La efectividad de los bloqueadores beta se documentó en un amplio estudio en el que se incluyó a 869 pacientes con SQTL, y se observó una reducción de los eventos cardiacos en los probandos de 0,97 a 0,31 episodios por año y en los familiares afectados, de 0,26 a 0,15 episodios por año29. En esta cohorte, con la excepción de la paciente V.1, no se produjeron eventos tras el inicio del tratamiento con bloqueadores beta, y se observó una reducción significativa del intervalo QTc.
El efecto protector de los bloqueadores beta en la población de pacientes con SQTL no es uniforme, posiblemente debido a que su mecanismo de acción está relacionado con la atenuación de los desencadenantes de carácter adrenérgico. Datos previos indicaban una alta tasa de eventos cardiacos entre los pacientes con QTL2 y SQTL tipo 3 tratados con bloqueadores beta30. Un estudio posterior publicado en 2011 sostiene que en la población con QTL2 la respuesta al tratamiento es específica del desencadenante, de tal manera que los pacientes con QT2 en tratamiento con bloqueadores beta presentan una tasa muy baja de episodios desencadenados por el ejercicio, pero alta tasa de eventos cardiacos desencadenados por el despertar (cambios de la frecuencia cardiaca más bruscos que con el ejercicio) y de eventos no desencadenados con el ejercicio o el despertar (no asociados a activación simpática)31. Sin embargo, un 13 y un 17% de los pacientes que experimentaron un primer evento desencadenado por el despertar o no desencadenado por el ejercicio ni el despertar, respectivamente, presentaron un episodio posterior en relación con el ejercicio32. Migdalovich et al5 describieron que el tratamiento médico con bloqueadores beta se asociaba a una reducción del 61% del riesgo de muerte súbita en una población de 1.166 pacientes con QTL2. Un estudio reciente32 en el que se comparó la eficacia de los bloqueadores beta de uso más frecuente mostró que los pacientes en tratamiento con propranolol presentaban un mayor acortamiento del QTc que los que recibían metoprolol y nadolol.
El único paciente que permaneció sintomático a pesar de los bloqueadores beta se sometió a denervación simpática cardiaca izquierda. En un estudio33 que incluyó a 147 pacientes con SQTL, la denervación simpática cardiaca izquierda redujo los eventos cardiacos en aproximadamente un 90%.
Estratificación del riesgoLa estratificación del riesgo más ampliamente aceptada para el SQTL se basa en las tasas de mortalidad publicadas y clasifica a los pacientes en riesgo alto (antecedentes de muerte súbita resucitada y/o taquicardia ventricular polimórfica documentada), intermedio (síncope previo y/o QTc > 500ms) y bajo (ausencia de síncope previo y QTc ≤ 500ms)29. Sin embargo, un estudio reciente mostró diferencias entre el tipo de mutación y las manifestaciones clínicas a lo largo del tiempo en los pacientes con QTL2. Así, tras el inicio de la adolescencia, las mujeres con y sin mutaciones de alto riesgo presentan aumento del riesgo de eventos cardiacos, mientras que en los varones el riesgo aumenta tan solo en los portadores de las mutaciones de alto riesgo que afectan al poro del canal5. En consecuencia, la tendencia actual para mejorar la estratificación del riesgo en el QTL2 es la evaluación combinada de los datos clínicos del paciente y las características específicas de la mutación.
CONCLUSIONESEste artículo describe por primera vez en una amplia familia de portadores las manifestaciones clínicas y los valores del intervalo QTc de la mutación missense KCNH2-H562R. En la familia descrita, se corroboran los datos previos acerca del alto riesgo asociado a las mutaciones del poro y se destaca la heterogeneidad clínica y electrocardiográfica de esta canalopatía. En este contexto, nuestro estudio resalta el papel fundamental que han adquirido los tests genéticos para decidir qué individuos se beneficiarían de tratamiento. Finalmente, con este artículo se resalta la importancia actual del análisis de cosegregación de la mutación con la enfermedad en la familia, especialmente en una época de desarrollo tecnológico en que las técnicas de secuenciación de nueva generación están adquiriendo un papel cada vez más importante.
CONFLICTO DE INTERESESNinguno.