Se presenta una familia con múltiples pacientes afectados de una forma clínica incompleta de síndrome de Marfan (SM), denominada fenotipo MASS. El fenotipo MASS hace referencia a pacientes con prolapso de la válvula mitral (M), dilatación aórtica (A) no progresiva y alteraciones cutáneas (S de skin) y osteomusculares (S de skeletal), similares al SM, pero sin cumplir los criterios de este1. El estudio genético familiar confirmó la patogenicidad de una variante no sinónima (NP_000129.3:p.Pro1424Ser, NM_000138.4:c.4270C>T) del gen de la fibrilina 1 (FBN1).
Los casos índice eran 2 hermanos intervenidos por dilatación de la raíz aórtica mediante sustitución aórtica (IV.8 y IV. 9 de la figura 1A). Referían antecedentes de muerte súbita de su tía y su madre, cuyas autopsias mostraron disección aórtica. Se realizaron estudios oftalmológico y del aparato locomotor, tomografía computarizada toracoabdominal y a 1 de ellos, estudio genético mediante secuenciación masiva dirigida con un panel de 30 genes. Este panel permitía analizar variantes puntuales y copy-number variation (CNV) (grandes deleciones y duplicaciones) y descartar otros síndromes diferenciales en un mismo estudio de manera coste-eficaz.

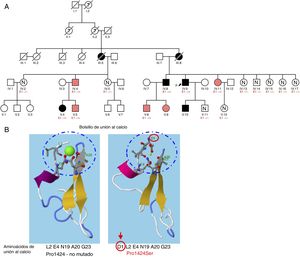
A: árbol genealógico; los sujetos en negro cumplen los criterios de síndrome de Marfan y en rojo, aquellos con fenotipo MASS. B: análisis in silico de bolsillo de unión al calcio del dominio EFG-like 24 mediante el programa RaptorX para comparar el aminoácido no mutado y el cambio que causaría Pro1424Ser en FBN1. E1–/+: heterocigoto para Pro1424Ser; E1–/–: no portador. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
El estudio genético detectó un cambio de sentido en una región codificante del exón 34 en FBN1 (cambio de un nucleótido por otro que originaba un cambio de aminoácido: p.Pro1424Ser). Esta variante se encontró en la UMD-FBN1 Mutation Database en un caso aislado que cumplía los criterios de SM, pero sin estudio de cosegregación familiar, imprescindible para catalogar una variante como patogénica1,2. No se detectó esta variante en ninguna otra base de datos de genotipificación pública. La búsqueda de esta variante en una base de datos poblacional como ExAC o gnomAD no identificó portadores, lo que indica una frecuencia alélica muy baja en poblaciones de control. Los estudios in silico con el programa RaptorX mostraron que el aminoácido prolina interviene en la formación de puentes de hidrógeno necesarios en el bolsillo de unión al calcio de esta región (epidermal growth factor [EGF] like 24). Su sustitución alteraría este bolsillo e implicaría un mal plegamiento proteico que alteraría su funcionalidad (figura 1B)3.
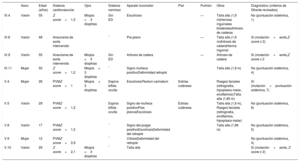
Además, se completó el estudio familiar (figura 1A). Se detectó la variante c.4270C>T en 9 familiares con expresión variable de signos de SM (tabla 1). Solo en los probandos y el caso V.10 se reportó dilatación aórtica, y el hallazgo cardiovascular más frecuente fue el prolapso valvular mitral. Dos pacientes presentaron miopía> 3 dioptrías y en ninguno se identificó ectopia lentis. Las alteraciones esqueléticas estaban presentes en prácticamente todos los pacientes. La gran mayoría mostraba escoliosis, pie plano o talla alta, aunque el signo de la muñeca o el pulgar solo se demostró en 3 pacientes. La evaluación de los demás pacientes no portadores no mostró hallazgos patológicos.
Características fenotípicas de la familia
| Sexo | Edad (años) | Sistema cardiovascular | Ojos | Sistema nervioso | Aparato locomotor | Piel | Pulmón | Otros | Diagnóstico (criterios de Ghante revisados) | |
|---|---|---|---|---|---|---|---|---|---|---|
| IV.4 | Varón | 55 | Z score=1,3 | Miopía <3 dioptrías | Sin ED | Escoliosis | — | Talla alta (1,9 m)Hernias inguinales bilateralesArtrosis de caderas | No (puntuación sistémica, 1) | |
| IV.8 | Varón | 48 | Aneurisma de aorta intervenido | * | Pie plano | — | Talla alta (1,9 m)Artrosis de caderaHernia inguinal | Sí (mutación+aorta,Z score ≥ 2) | ||
| IV.9 | Varón | 55 | Aneurisma de aorta intervenido | Miopía <3 dioptrías | Sin ED | Artrosis de cadera | — | Artrosis de cadera | Sí (mutación+aorta,Z score ≥ 2 | |
| IV.11 | Mujer | 50 | Z score=1,2 | Miopía> 3 dioptrías | * | Signo muñeca positivoDeformidad retropié | Talla alta (1,8 m) | No (puntuación sistémica, 4) | ||
| V.4 | Mujer | 26 | PVMZ score=1 | Miopía> 3 dioptrías | Espina bífida oculta | EscoliosisPectum carinatum | Estrías cutáneas | Rasgos faciales (retrognatia, hipoplasia malar, enoftalmos)Talla alta (1,85 m) | Sí (mutación+puntuación sistémica, 7) | |
| V.5 | Varón | 29 | PVMZ score=1,2 | Espina bífida oculta | Signo de muñeca positivoPies planosEscoliosis | Estrías cutáneas | Talla alta (1,9 m), Rasgos faciales (retrognatia, enoftalmos, hipoplasia malar) | No (puntuación sistémica, 6) | ||
| V.8 | Varón | 17 | PVMZ score=1,3 | * | Signo del pulgar positivoEscoliosisDeformidad del retropié | Talla alta (1,96 m) | No (puntuación sistémica, 5) | |||
| V.9 | Mujer | 12 | PVMZ score=0,9 | * | CifosisDeformidad del retropié | No (puntuación sistémica, 4) | ||||
| V.10 | Varón | 29 | Z score=2,1 | Miopía <3 dioptrias | * | Talla alta | Sí (mutación+aorta, Z score ≥ 2) |
ED: ectasia dural; PVM: prolapso de válvula mitral.
Estos datos en conjunto indican una cosegregación familiar de la variante con formas clínicas variables, tal como se ha descrito previamente con variantes patogénicas en este gen4. Por ello, es importante realizar estudios familiares completos en centros de referencia para obtener un mayor conocimiento del comportamiento fenotípico de las variantes5. Salvo los sujetos V.4, IV.8 y IV.9, el resto de los pacientes pueden catalogarse como de fenotipo MASS al no cumplir alguno de los criterios revisados de SM de Ghent1. Puede que la juventud de gran parte de los portadores influya en la presencia o ausencia de manifestaciones cardinales como dilatación aórtica o ectopia lentis. Se ha postulado que variantes en trans en FBN1 pudieran tener un papel modificador4. Recientemente, un estudio ha demostrado la presencia de variantes reguladoras de expresión génica en el mismo alelo que la variante patogénica, lo que indicaría que esta interacción podría explicar la penetrancia incompleta y la expresividad variable en portadores6. Aunque no se detectó ectasia dural en los portadores, cabe destacar la presencia de espina bífida oculta en 2 hermanos (V.4 y V.5).
Se ha descrito otra variante que afectaría al mismo aminoácido Pro1424Ala (g.48764814G>C), identificada en diferentes individuos con SM. Sin embargo, no se dispone de estudios de cosegregación y consta identificada en 54/277.166 individuos de poblaciones de control del gnomAD, por lo que su patogenicidad no está aclarada.
En conclusión, el estudio clínico y genético de esta familia ha permitido determinar la asociación de la variante p.Pro1424Ser en FBN1 con el desarrollo de fenotipo MASS y el SM. Si bien la variabilidad fenotípica causada por dicha variante es amplia con casos de afección leve, es importante la monitorización seriada de diámetros aórticos de los portadores, pues puede asociarse con aneurisma y disección.
CONFLICTO DE INTERESESJ.P. Trujillo-Quintero y L. Montserrat forman parte de la empresa de diagnóstico genético Health in Code.