

En los últimos años, la genética ha adquirido merecidamente un lugar importante en casi todas las disciplinas médicas, y este también es el caso en el campo de las cardiopatías congénitas. Esto no solo ha llevado a una mejor comprensión de la fisiopatología de los defectos cardiacos congénitos, sino que también conlleva un impacto positivo en el tratamiento del paciente. La integración de la genética clínica en centros acreditados para el abordaje de las cardiopatías congénitas es sin duda una recomendación clara. Los cardiólogos pediátricos y de adultos tienen un papel crucial en el proceso de evaluación genética de los pacientes y sus familias, por lo que deben conocer las señales de alerta que justifiquen un estudio genético más o menos elaborado, así como el asesoramiento y la realización de otras pruebas. Para la correcta interpretación de los resultados de las pruebas genéticas, es esencial disponer de algunos conocimientos básicos. En este documento de revisión se proporciona una visión general práctica de lo que implica la evaluación genética, qué tipo de pruebas genéticas son posibles hoy y cómo se aplican al paciente individual en la práctica clínica.
Palabras clave
Las cardiopatías congénitas (CPC) se encuentran entre los defectos neonatales más frecuentes y afectan aproximadamente a un 1% de los niños nacidos vivos1. En los últimos años se han hecho avances importantes tanto en la exactitud del diagnóstico clínico como en el tratamiento de las CPC. Ello ha prolongado la supervivencia y ha mejorado la calidad de vida de una gran parte de los pacientes afectados, de tal manera que hoy el número de individuos adultos con CPC supera al de niños afectados en muchos países2. Junto con este avance, la demanda y la búsqueda de posibles explicaciones a la causa subyacente a los defectos cardiacos han aumentado en los últimos años, tanto por los pacientes y los padres como por los profesionales de la salud involucrados. Los recientes avances tecnológicos han impulsado nuestro conocimiento del fundamento genético de las formas sindrómicas de CPC. Sin embargo, en la CPC sola, hay una gran falta de conocimiento de los mecanismos moleculares, si bien varias líneas de evidencia indican que la genética contribuye a ello de manera importante: la incidencia de las CPC que afectan a ambos gemelos es mayor en los monocigotos que en los dicigotos3, el riesgo de recurrencia de la CPC de hermanos e hijos de pacientes con CPC es mayor que para la población general4,5 y el empleo de tecnologías de análisis masivo de gran capacidad permite la identificación de una anomalía genética en hasta una tercera parte de los pacientes con CPC si se consideran conjuntamente las formas sindrómicas y no sindrómicas6.
El conocimiento de la causa o las causas genéticas subyacentes será útil para realizar un ajuste fino personalizado del asesoramiento y las opciones de tratamiento. En diversas alteraciones ya se ha establecido que un defecto genético subyacente influye en el tratamiento y los resultados de la CPC. Por ejemplo, se debe vigilar cuidadosamente a los pacientes con una comunicación interauricular (CIA) debida a variantes patógenas de NKX2.5 para detectar posibles arritmias7. Además, en especial en los niños, el defecto subyacente puede indicar un riesgo de complicaciones no cardiacas, como retraso del desarrollo neurológico, problemas respiratorios o disfunción renal, que requieran una intervención temprana o un seguimiento para prevenir o aliviar esas manifestaciones.
Por último, el conocimiento de la base genética influirá en el asesoramiento sobre el riesgo de recurrencia de hermanos e hijos y puede dar acceso a opciones de reproducción mediante el diagnóstico genético prenatal y preimplantacional. Durante las últimas décadas, la integración de los datos epidemiológicos, clínicos y genéticos ha mejorado significativamente el conocimiento existente sobre la recurrencia de las CPC. La hipótesis predominante de que la CPC se hereda en forma de rasgo multifactorial se puso en duda ya en la década de los ochenta8. Rose et al.9 observaron una frecuencia de CPC superior a la esperada en función de lo indicado por modelos multifactoriales en varias familias. El mayor uso y el perfeccionamiento de las técnicas de imagen condujeron a la importante observación de que algunas alteraciones (aunque no todas) pertenecen a un espectro fenotípico de CPC más amplio que puede darse en un contexto familiar. Entre los ejemplos bien establecidos, se encuentran los defectos del tracto de salida del ventrículo izquierdo (TSVI): se observó que los familiares de niños con un síndrome de hemicardio izquierdo hipoplásico o una obstrucción del TSVI presentaban una tasa de válvula aórtica bicúspide (VAB) superior a la esperada10,11. Además de lo mencionado, conocer la causa subyacente puede tener un efecto «terapéutico», y ayudar a los familiares a afrontar y aceptar una enfermedad rara.
Las nuevas técnicas genéticas basadas en análisis a gran escala, con tiempo de obtención corto y costes accesibles, han aumentado la disponibilidad del diagnóstico genético. Se prevé que esta tendencia continúe a un ritmo rápido y que la genética proporcione respuestas en un número creciente de casos.
No obstante, un análisis genético detallado implica también dificultades crecientes de interpretación de los resultados y un abanico en constante evolución de posibilidades e inconvenientes. Esta situación es la que ha motivado esta revisión sobre la situación actual de las pruebas genéticas en el campo de las CPC.
FUNDAMENTOS GENÉTICOSDefinición de la genética¡No realizar pruebas sin acompañarlas de asesoramiento!Al definir la «genética», es importante establecer una distinción entre los conceptos de «asesoramiento genético» y «pruebas genéticas». Es necesario resaltar desde el principio que ambos conceptos están intrínsecamente ligados (las pruebas genéticas deben acompañarse siempre de un asesoramiento correcto), pero que el asesoramiento no en todos los casos lleva a la realización de pruebas.
Según la Organización Mundial de la Salud, el asesoramiento genético se define como «el proceso a través del cual profesionales adecuadamente capacitados comparten el conocimiento sobre aspectos genéticos de enfermedades con las personas que presentan un aumento del riesgo de padecer un trastorno heredable o de transmitírselo a sus hijos antes de que nazcan».
El asesoramiento genético en el contexto de las CPC se introdujo hace ya más de medio siglo, cuando el contexto más importante de su uso era el de informar a los padres de un niño afectado sobre el riesgo de recurrencia. En los primeros estudios se demostró de manera impecable que informar a los padres en un proceso de asesoramiento específico tenía efectos beneficiosos12. La investigación más reciente ha confirmado los efectos beneficiosos de las sesiones individualizadas de asesoramiento genético para los padres de niños con CPC por lo que respecta a la mejora del conocimiento acerca de las causas de la CPC y de la funcionalidad psicosocial, lo cual hace que se recomiende claramente su inclusión en la práctica clínica habitual13. Con un número de adultos con CPC en aumento, las indicaciones para el asesoramiento genético y las pruebas genéticas se han ampliado y la «genética» es un requisito que ha pasado a formar parte de los programas de CPC de adultos en la guía recientemente publicada por el American College of Cardiology y la American Heart Association sobre las CPC del adulto14.
Los asesores genéticos en CPC son profesionales de la salud con formación de grado universitario que han recibido una capacitación específica tanto en genética médica como en asesoramiento, centrada en especial en las CPC. Los asesores genéticos elaboran un árbol genealógico de 3 generaciones y recogen todos los datos clínicos pertinentes del probando y sus familiares, prestando especial atención a los abortos y las muertes neonatales. Además de su papel en la obtención de los antecedentes familiares y el asesoramiento de los pacientes y las familias acerca del riesgo de recurrencia, el riesgo de un síndrome concreto y la interpretación de los resultados, los asesores genéticos pueden desempeñar un papel importante en la clasificación de los pacientes que se debe remitir a una evaluación genética completa15.
El estudio genético de las CPC requiere un abordaje multidisciplinario en el que, además del cardiólogo (pediátrico) y el asesor genético, es también crucial el genetista clínico. Los genetistas clínicos son médicos que han recibido una capacitación específica en la evaluación diagnóstica, el tratamiento y el asesoramiento genético. Los programas de capacitación y la certificación son específicos para cada país. Los genetistas clínicos determinarán si el defecto cardiaco es único o forma parte de un síndrome, lo cual es necesario para orientar las pruebas genéticas y determinar el abordaje médico. Según lo indicado por estudios epidemiológicos grandes, las malformaciones cardiovasculares sindrómicas constituyen como mínimo un 25% del total de malformaciones cardiovasculares4,16. La investigación realizada en el contexto de la deleción de 22q11ha puesto ya de manifiesto que los cardiólogos alcanzan un menor rendimiento en la evaluación de los síndromes, por lo que la evaluación de genética clínica es deseable17. Una vez se ha determinado si un paciente tiene una entidad sindrómica o no, el tratamiento de las formas sindrómicas también puede estar mejor coordinado por un genetista clínico en el contexto de un equipo multidisciplinario; naturalmente, es mejor que el seguimiento de los pacientes con formas de CPC aisladas lo haga el cardiólogo (pediátrico)8.
Otra cuestión importante que tener en cuenta en el proceso del asesoramiento genético y las pruebas genéticas es el consentimiento. Para cualquier prueba genética es esencial que el paciente (o su representante legal) conozca los beneficios y los riesgos de esa prueba y dé su consentimiento por escrito para realizarla. Queda fuera de lo abordado en este artículo el análisis de aspectos (importantes) como los hallazgos casuales y las pruebas presintomáticas, pero quisiéramos abordar brevemente las pruebas dirigidas directamente al consumidor (DTC).
En muchos países han quedado ya atrás los días en que las pruebas genéticas se realizaban tan solo en centros de genética clínica con certificación (pruebas dirigidas por el laboratorio), en los que se aplicaban reglas estrictas para la realización de las pruebas clínicas y moleculares. Como resultado, por un lado, de los avances técnicos que se han producido en las pruebas genéticas y, por otro, de la creciente demanda pública, se ha asistido a un aumento significativo del número de empresas que ofrecen pruebas conocidas como DTC. Se trata de pruebas en las que las muestras (de sangre o saliva) se envían por correo directamente al laboratorio, sin un asesoramiento previo. Las pruebas genéticas DTC pueden detectar trastornos monogénicos graves y de penetrancia elevada o variantes genéticas asociadas con un aumento de la susceptibilidad a enfermedades frecuentes y complejas. Preocupa la posibilidad de que la interpretación de variantes basada en pruebas DTC no siempre sea correcta. Han aparecido ya presentaciones de casos de tratamiento innecesario de familiares sanos o de tranquilización errónea basada en una información incorrecta18. En un estudio se observó que un 40% de las variantes de diversos genes indicadas por los datos brutos de pruebas DTC correspondían a falsos positivos19. Esta información falsa tiene repercusiones graves, en primer lugar, en los pacientes y sus familias, pero también implica una sobrecarga de los servicios de asesoramiento genético a los que se consulta para esclarecer y rectificar los resultados de pruebas solicitadas en otros lugares20. Ni que decir tiene que estas cuestiones crean tensión en el contexto de las pruebas genéticas DTC por lo que respecta a las expectativas y la evaluación normativa de las estrategias de comunicación21.
Por estas razones, la European Society of Human Genetics ha establecido una política sobre la publicidad y la prestación de servicios de pruebas genéticas predictivas por estas empresas de DTC22. Nosotros estamos en contra del uso de pruebas DTC en el ámbito de las pruebas genéticas de CPC.
La (r)evolución técnica de las pruebas genéticasEvolución en la citogenéticaEl hito que supuso el descubrimiento de la trisomía 21 como causa genética del síndrome de Down en 195923 introdujo las pruebas genéticas en las CPC. Desde entonces, las nuevas metodologías y el ajuste fino de las técnicas han conducido a la identificación de la causa genética de las CPC (principalmente en las formas sindrómicas). La determinación clásica del cariotipo con el patrón de bandas G tiene una resolución bastante baja, de 3-5 Mb, y actualmenet solo se realiza para indicaciones específicas, como la confirmación de aneuploidías (mosaicismo) (síndrome de Down, síndrome de Turner, mosaicismo de trisomía 8) y translocaciones familiares.
La hibridación in situ fluorescente (FISH) utiliza una sonda con marcación fluorescente dirigida a regiones genómicas específicas. Se emplea para la detección dirigida de aberraciones que se encuentran por debajo de la resolución que proporciona el cariotipo, como las microdeleciones de 22q11.2 o 7q11.2 en el síndrome velocardiofacial y el síndrome de Williams-Beuren respectivamente. Un importante avance fue la introducción de la hibridación genómica comparativa matricial (ArrayCGH)24, también denominada análisis de micromatrices o microchips cromosómicos. El análisis de microchip cromosómico utiliza una hibridación competitiva de ADN desmenuzado de control y ADN del paciente marcado con diferentes fluorocromos en una matriz que contiene decenas de miles de sondas moleculares dispersas en todo el genoma de referencia. A continuación, una lectura automática de las diferencias en la intensidad del color detecta en todo el genoma las variaciones en el número de copias (CNV), es decir, deleciones o duplicaciones de tan solo 100 kb. Esta técnica se denomina también «determinación molecular del cariotipo». La matriz o chip de polimorfismo de un solo nucleótido (SNP) es una prueba similar que utiliza los SNP para detectar regiones con una pérdida de la heterocigosidad. Sin embargo, la enorme variabilidad estructural en el genoma humano ha dificultado una interpretación sencilla de los resultados de las pruebas, especialmente en los años siguientes a la introducción de la prueba en el ámbito diagnóstico. Iniciativas como la Database of Genomic Variants25 han tenido efectos negativos en documentar la variación normal, mientras que bases de datos como DECIPHER26 han desempeñado un papel prominente en la identificación de nuevos defectos estructurales genómicos como base de la enfermedad. La determinación molecular del cariotipo introdujo el concepto de «genética inversa», una estrategia mediante la cual se compara a pacientes con la misma variante genética para identificar una correlación de genotipo-fenotipo y delimitar nuevas entidades clínicas, como el síndrome de Koolen-de Vries27. A continuación se detallan las variantes estructurales asociadas más frecuentes en las CPC que se conocen hasta la fecha. Más recientemente, el análisis de microchip cromosómico está siendo reemplazado por métodos basados en tecnologías de secuenciación genómica de baja cobertura («superficiales») (véase más adelante).
Evolución en la genética molecularEl uso combinado de la reacción en cadena de la polimerasa y la secuenciación Sanger introdujo el análisis molecular en las consultas clínicas. No obstante, el análisis era costoso y laborioso. Además, la identificación de nuevos genes causantes de fenotipos cardiovasculares estaba limitada a las formas sindrómicas que se podía estudiar mediante análisis de linkage en familias grandes con una herencia dominante del trastorno (p. ej., síndrome de Noonan28), o en familias con consanguinidad que tenían trastornos recesivos (p. ej., Ellis-van Creveld29), mientras que los enfoques de gen candidato tan solo identificaban esporádicamente un defecto casual, a menudo con la ayuda de la detección previa de una microdeleción que incluía el gen candidato (p. ej., síndrome CHARGE30). Un segundo avance fue el que se produjo con la introducción de la secuenciación de nueva generación (NGS) (o paralela masiva)31. Resumidamente, en la NGS, los fragmentos de ADN de la región o las regiones de interés (ya sea un panel específico de genes, el exoma, es decir, las regiones codificadoras del ADN, o el genoma, es decir, la secuencia completa del ADN) se secuencian en paralelo, y las «lecturas» obtenidas se alinean con la secuencia de referencia. Se denomina cobertura en una determinada posición del genoma al número de veces que una base en una posición genómica determinada es secuenciada de manera independiente. Para una interpretación fiable, es necesaria una cobertura de como mínimo 20 veces. Los métodos de secuenciación de lectura corta se emplean principalmente en laboratorios clínicos debido a su relación de coste-efectividad y la baja tasa de errores por base. Con ello, la aplicación de la secuenciación del exoma puede facilitar el análisis de un número amplio de genes que se sabe que causan CPC o incluso identificar nuevos genes candidatos para CPC. Sin embargo, las longitudes de lectura cortas (50–500pb) pueden producir alineaciones erróneas y ensamblajes erróneos en zonas de gran complejidad del genoma; no pueden cubrir repeticiones de una manera viable y causan un deterioro de las fases de las variantes. Además, el proceso de amplificación, que es indispensable en la secuenciación de lectura corta, crea una infrarrepresentación de bases en zonas con un contenido alto o bajo de guanina-citosina (GC).
De nuevo, dada la enorme variabilidad del genoma humano, la interpretación de variantes es crucial, fundamentada en las bases de datos de libre acceso y la predicción de causalidad mediante herramientas de bioinformática (véase más adelante). De manera similar a DECIPHER, surgieron bases de datos como GeneMatcher32 para catalizar el descubrimiento de enfermedades raras aportando una plataforma para conectar a los clínicos y los investigadores de todo el mundo que tenían un interés común en el mismo gen.
Secuenciación de tercera generación: ¿evolución hacia una única prueba genómica?A pesar de que el análisis del número de copias (con una resolución de como mínimo 100kb) y el análisis de secuencia de alto rendimiento emplean NGS de lectura corta, la mayor parte de la variación estructural continúa sin detectarse. Las variantes estructurales, que incluyen CNV, inversiones y translocaciones, constituyen un 10% del genoma y contribuyen a producir una diversidad entre 2 genomas humanos mayor que la que causa ninguna otra forma de variación genética y pueden afectar a la expresión de los genes.
Además de los defectos cromosómicos grandes como las translocaciones, las inversiones y en especial las CNV, las variantes estructurales crípticas más pequeñas (de entre 50pb y 50kb) pueden causar también enfermedades en el ser humano al afectar a la función o la expresión de los genes; por ejemplo, las variantes estructurales> 20 kb tienen una probabilidad de afectar a la expresión de un gen hasta 50 veces mayor que la de un SNV.
Las plataformas de secuenciación del genoma de tercera generación utilizan una secuenciación de lectura larga (LRS) (> 10 kb), permiten determinar variaciones estructurales con una resolución sin precedentes y superan la mayoría de las limitaciones de la secuenciación de lectura corta33. Con el tiempo, esto conducirá a un análisis único para cubrir la mayor parte de la variación del ADN del genoma en un futuro próximo.
La secuenciación genómica de tercera generación puede combinarse con otros métodos de NGS, como la secuenciación de transcriptoma (biblioteca de genes expresados en un determinado tipo de célula) con objeto de identificar la variación a nivel de expresión y de corte y empalme. No obstante, la principal dificultad continuará siendo la interpretación de las variantes, que a menudo requiere una validación adicional y continuará siendo la carga principal de la aplicación diagnóstica de estas técnicas (véase más adelante).
Interpretación de los resultados de las pruebas genéticasNecesidad de tratamiento y conservación de datos de genesCon los avances técnicos de las pruebas genéticas antes descritos, y que ahora permiten analizar simultáneamente un gran número de genes, ha habido una gran tentación de crear paneles más amplios para trastornos específicos. Los laboratorios genéticos comerciales en especial han seguido este camino, y actualmente ofrecen paneles para CPC que contienen, por ejemplo, más de 100 genes.
Sin embargo, se recomienda cierta precaución en esta tendencia. Para múltiples trastornos, hay evidencia sólida de que el análisis de más genes conduce inevitablemente a la detección de más «variantes de significado desconocido» (VUS), cuya interpretación no resulta fácil y a veces conlleva incluso el riesgo de generar a los pacientes una ansiedad y una discriminación innecesarias34,35. Al seleccionar los genes que incluir en los paneles diagnósticos o la notificación de la secuenciación del exoma/genoma, es necesario considerar cuidadosamente la validez clínica, es decir, la solidez de la evidencia que indica que una modificación en ese gen predispone a la enfermedad. Clinical Genome Resource o ClinGen han elaborado un marco de referencia para la evaluación semicuantitativa de la validez de la asociación de gen y enfermedad para muchas enfermedades (pero aún no para la CPC). En este marco de referencia, los genes se clasifican en niveles preestablecidos basados en la evidencia clínica, genética y experimental, junto con la valoración y el consenso de expertos del ámbito clínico. Estos genes validados clínicamente pueden usarse para establecer prioridades en qué genes estudiar y para aportar información a la hora de decidir qué genes deben incluirse en los paneles para enfermedades36,37.
Necesidad de tratamiento y conservación de datos de variantesLa posibilidad de generar grandes cantidades de datos genéticos a través de un examen de detección genética amplio ha permitido establecer un diagnóstico genético rápido y más exacto. Sin embargo, como ya se ha mencionado, cada análisis proporciona múltiples variantes genéticas (hasta 50.000 variantes por exoma), y ello requiere una interpretación apropiada. Es esencial diferenciar las variantes benignas de las patógenas para trasladar los resultados genéticos a la práctica clínica, pero esto sigue siendo un auténtico reto. Además, el número de VUS continúa siendo demasiado elevado. En 2015 el American College of Genetics and Genomics (ACMG) y la Association for Molecular Pathology (AMP) propusieron una guía para clasificar las variantes genéticas relacionadas con trastornos mendelianos38. La clasificación se basa en 28 criterios para considerar finalmente una variante como benigna (clase 1), probablemente benigna (clase 2), VUS (clase 3), probablemente patógena (clase 4) o patógena (clase 5); las clases 2 y 4 implican una certeza> 90% de que una variante sea benigna o causante de enfermedad. Los criterios incluyen características clínicas, datos obtenidos de grandes bases de datos de exoma humano, como gnomAD y ExAC, y datos que abordan el efecto estructural de una variante en el ADN o las proteínas.
Son necesarios datos clínicos correctos y detallados no solo del probando, sino a menudo también de familiares. Así, la cooperación con (y de) los familiares es de gran importancia, tanto en el contexto diagnóstico como en la posterior comunicación de los resultados. Una subdivisión más detallada de las variantes identificadas (variantes del número de copias y variantes de un solo nucleótido) requiere una verificación en familiares de primer grado, en los que, además de los estudios del ADN, es preciso también un examen clínico cardiovascular, con objeto de evaluar adecuadamente la presencia o ausencia de anomalías clínicas en el individuo. Es importante comunicar esto al paciente de modo apropiado desde el inicio del proceso de asesoramiento y realización de pruebas.
Si se confirma un resultado anormal de la prueba, se recomienda también verificar el resultado nuevamente con otros familiares. Por ley, no se permite a los cuidadores contactar con familiares; el contacto debe hacerse a través del paciente (o su representante) y también es importante comunicar esto correctamente al paciente cuando se comentan los resultados.
Además de los criterios clínicos en pacientes y familiares, se tienen en cuenta también las características moleculares de las variantes. El análisis computacional de una variante con modelización de los efectos esperados del gen o la variante en las estructuras y la función proteicas puede aportar evidencia de apoyo para establecer la patogenicidad. A pesar de una interpretación cuidadosa y de las iniciativas internacionales respecto al tratamiento y conservación de los datos, las consecuencias clínicas de muchas de las variantes obtenidas a través de un análisis molecular en profundidad continúan siendo desconocidas. Hay varias herramientas, como el análisis transcriptómico, proteómico, metabolómico, lipidómico y meilómico, que pueden ser útiles para facilitar la identificación de las consecuencias moleculares. Sin embargo, para la mayoría de los genes que son defectuosos en la CPC, las respectivas firmas (multi)ómicas no se conocen. Además, es posible que algunos genes solamente tengan trascendencia durante el desarrollo cardiaco, de tal manera que las pruebas posnatales en otros tejidos podrían ser irrelevantes. Por otra parte, la elaboración de modelos animales para enfermedades específicas es laboriosa, costosa e imposible en el contexto clínico. Sin embargo, la mutagénesis directa con técnicas de CRISPR_Cas9 está pasando a ser cada vez más eficiente y es posible que llegue a ser útil en la interpretación de las variantes genómicas.
Aunque la guía de ACMG/AMP introdujo de manera clara algunas mejoras importantes en la interpretación de las variantes genéticas, a menudo continúa dejando mucho espacio a la interpretación subjetiva y, por consiguiente, varios grupos han propuesto clasificaciones genéticas más específicas39.
Tras la publicación de la guía, se han elaborado varias herramientas para facilitar la interpretación de las variantes genéticas (tabla 1). Además, se dispone también de varios repositorios online en los que pueden consultarse las variantes ya clasificadas anteriormente por otros laboratorios (tabla 1). No obstante, debe tenerse precaución al consultar estos repositorios, ya que la interpretación de las variantes continúa siendo subjetiva y no siempre la han realizado grupos con experiencia. Clinvar, que es uno de estos archivos, no solo proporciona una interpretación de una variante específica, sino que aporta también un nivel de revisión para respaldar la afirmación de trascendencia clínica.
Cuadro general de las herramientas de acceso libre online para la clasificación e interpretación de las variantes genéticas
| Herramientas para la clasificación de las variantes genéticas | |
|---|---|
| Herramienta | Descripción |
| Clingen Pathogenicity Calculator40,41 | Basada en las guías de ACMG/AMP y aportaciones adicionales de expertosRequiere entrada de datos manualRequiere registroOpción de envío directo a Clinvar |
| Varsome42,43 | Basada en las guías de ACMG/AMP; sin aportaciones de expertos (?), pero con apoyo bioinformático adicionalInterpretación automática de variantesOpción de modificación manual de la clasificación |
| Intervar44,45 | Basada en las guías de ACMG/AMP, ¿sin aportaciones de expertos?Interpretación automática de variantesOpción de modificación manual de la clasificación |
| Franklin46 | Basada en las guías de ACMG/AMP, ¿sin aportaciones de expertos?Interpretación automática de variantesOpción de modificación manual de la clasificación |
| Cardioclassifier47,48 | Tan solo para enfermedades cardiovascularesBasada en las guías de ACMG/AMP y algún conocimiento experto específicoInterpretación automática de variantes, sin opción de modificación manual de la clasificaciónRequiere registro gratuito |
| Repositorios online | |
|---|---|
| Clinvar49,50 | Asociado a ClinGenLa clasificación es examinada por expertos y se le asigna una categoría revisadaActualización periódica |
| Leiden Open Variation Database51,52 | Asociada al Human Variome ProjectActualización periódica de las variantes clasificadas |
| Universal Mutation Database53,54 | Asociada a la Human Genoma Variation SocietyDatos limitados a ciertas variantes con especificidad de locus |
| Human Gene Mutation Database55,56 | Requiere registro gratuitoRepositorio de los principales datos publicados sobre una variante determinada, en vez de un archivo de interpretaciónActualización periódica |
ACMG/AMP: American College for Medical Genetics/Association for Molecular Pathology.
La genética es un campo que evoluciona dinámica y rápidamente. Para muchos de los fenotipos clínicos se están descubriendo periódicamente nuevos datos genómicos, y es posible que los resultados de las pruebas obtenidos hoy queden desfasados mañana. Por consiguiente, debe aplicarse una reconsideración regular y cuidadosa del asesoramiento genético y las pruebas genéticas, en especial en los individuos/familias con un alto grado de sospecha pero sin identificación de un defecto genético. En esta misma línea, la interpretación de una variante genética puede cambiar con el paso del tiempo y se debe revaluar periódicamente las variantes genéticas descubiertas antes (en especial las VUS) a la luz de los nuevos datos publicados57.
De nuevo, y en mayor medida aún que en el proceso de asesoramiento inicial, la repetición de las pruebas y la comunicación de los resultados alterados deben realizarse con precaución para asegurarse de que los pacientes y sus familias interpretan correctamente los resultados58.
EL ESPECTRO DE LOS DEFECTOS GENÉTICOS EN LA CPCHasta un 25-30% de los pacientes con CPC presentan otras manifestaciones extracardiacas asociadas59. Está bien establecida la asociación entre la CPC en varias aneuploidías cromosómicas y algunas CNV, como el síndrome de Down, el síndrome de Turner y el síndrome de deleción de 22q11. Otras CNV y variantes de un solo gen han mostrado también una penetrancia elevada en las CPC. Para las demás CPC no sindrómicas, se han identificado varios genes que muestran una herencia mendeliana (en su mayor parte autosómica dominante, pero en algunos casos también autosómica recesiva). Es de destacar que algunos de estos genes podrían estar involucrados tanto en casos sindrómicos como en casos no sindrómicos. En la tabla 2 y la tabla 3 se presenta un resumen de varias formas de CPC asociadas con trastornos genéticos, así como de las manifestaciones clínicas de mayor importancia en los síndromes más frecuentes. En términos generales, muchos genes corresponden a factores de transcripción, vías de señalización o remodeladores de la cromatina. Por consiguiente, el número de copias de un gen y la alteración de la expresión génica son un mecanismo de probable importancia en la CPC. En consecuencia, otros mecanismos, como variantes estructurales, actualmente podrían estar infradiagnosticados en las CPC. Además, la alteración en el número de copias génicas en fases cruciales del desarrollo crea una ventana temporal en la que factores ambientales pueden interferir en el desarrollo cardiaco. Por último, el mosaicismo somático en las células progenitoras cardiacas sigue siendo objeto de controversia.
Cuadro general de los diferentes genes y síndromes asociados con cardiopatías congénitas
| Cardiopatía congénita | Genes asociados (formas no sindrómicas) | Síndromes asociados |
|---|---|---|
| RVP(T)A | Síndrome de Turner (monosomía X)Síndrome de Kabuki. (MLL2, KDM6A) | |
| CIA | MYH6, ACTC1, GATA4, TBX20, TLL1, NKX2.5, CITED2, GATA6, TBX5 | Síndrome de Down (trisomía 21)Síndrome de deleción de 1p36Síndrome de Holt-Oram (TBX5)Síndrome de Ellis-van Creveld (EVC*)Síndrome de Kabuki (MLL2, KDM6A)Rasopatías (PTPN11, HRAS) |
| CIV | GATA4, NKX2.5, CITED2, TBX5, ETS1 | Síndrome de DownSíndrome de deleción de 22q11Síndrome de deleción de 1p36Síndrome de Jacobsen (deleción de 11q terminal)Síndrome de Holt-Oram (TBX5)Síndrome de Kabuki (MLL2, KDM6A)Síndrome de Ellis-van Creveld (EVC*)Rasopatías (PTPN11, HRAS) |
| CAV | GJA1*, GATA6, GATA4, CRELD1, NR2F2 | Síndrome de Down (trisomía 21) |
| SCIH | GJA1*, NKX2.5 | Síndrome de Turner (monosomía X)Síndrome de Jacobsen (deleción de 11q terminal)Síndrome de Adams-Oliver (NOTCH1)Síndrome de Kabuki (MLL2, KDM6A)Síndrome CHARGE (CHD7)Deleción de 22q11.2 |
| TF | NKX2.5, GATA4, GATA6, TBX1, JAG1, ZFPM2 | Síndrome de Down (trisomía 21)Síndrome de deleción de 22q11Síndrome de deleción de 1p36Síndrome CHARGE (CHD7)Síndrome de Kabuki (MLL2, KDM6A)Síndrome de Alagille (JAG1, NOTCH2)Síndrome de Myhre (SMAD4) |
| TGV | Síndrome de deleción de 22q11Síndrome de Jacobsen (deleción de 11q terminal)Discapacidad intelectual relacionada con MED13L | |
| Tronco arterioso | NKX 2.5, NKX2.6, GATA6, TBX1, ACTA2, R187 | Síndrome de deleción de 22q11Síndrome CHARGE (CHD7) |
| Coartación de aorta/cayado aórtico interrumpido | NKX 2.5, NKX2.6, GATA6, TBX1 | Síndrome de Turner (monosomía X)Síndrome de deleción de 22q11Síndrome CHARGE (CHD7)Síndrome de Myhre (SMAD4) |
| Anomalías de la válvula aórtica | NOTCH1, SMAD6, ROBO4 | Síndrome de Turner (monosomía X)Síndrome de Jacobsen (deleción de 11q terminal)Síndrome de Adams-Oliver (NOTCH1)Síndrome de Kabuki (MLL2, KDM6A) |
| Anomalía de Ebstein | GATA4, NKX2.5, MYH7 | Síndrome de deleción de 1p36Síndrome CHARGE (CHD7)Asociación VACTERLSíndrome de Kabuki (MLL2, KDM6A)Síndrome de Holt-Oram (TBX5)Síndrome de Cornelia de Lange |
| Anomalía de la válvula pulmonar | GATA4 | Rasopatías (PTPN11, HRAS) |
| Estenosis aórtica y pulmonar supravalvular | ELN | Síndrome de Williams-Beuren (deleción de 7q11.2)Síndrome de Alagille (JAG1, NOTCH2) |
| PDA | PRDM6, ACTA2, R187, MYH11 | Síndrome de Char (TFAP2B)Síndrome de deleción de 1p36 |
CAV: comunicación auriculoventricular; CIA: comunicación interauricular; CIV: comunicación interventricular; PDA: persistencia del ductus arteriosus; RVPA(T): retorno venoso pulmonar anormal (total); SCIH: síndrome de corazón izquierdo hipoplásico; TF: tetralogía de Fallot; TGV: transposición de grandes vasos.
Cuadro general de las principales manifestaciones sistémicas y cardiovasculares de los síndromes genéticos asociados con CPC
| Síndrome | Diagnóstico molecular | Principales manifestaciones sistémicas | Principales manifestaciones cardiovasculares |
|---|---|---|---|
| Aneuploidía cromosómica | |||
| Síndrome de Down | Trisomía 21Translocación del cromosoma 21Mosaicismo | Características faciales típicasDiscapacidad intelectualHipotonía, baja estaturaAtresia gastrointestinal | Frecuente: CAV, CIV, CIAOtros defectos asociados: PDA, TF |
| Síndrome de Turner | Mosaicismo monosomía X | Cuello alado y límite bajo de inserción posterior del peloLinfedemaBaja estatura, tórax en tonelPubertad retardadaInfertilidadPérdida de audición, problemas de ORLHepatopatía | Frecuente: VAB, CoA, dilatación de aorta ascendenteOtros defectos asociados: RVPA(T), SCIH |
| CNV | |||
| Síndrome 22q11 | Deleción de 22q11.2 | Características faciales típicas, habla nasalAnomalías palatinas y problemas de alimentaciónDificultades de aprendizajeInmunodeficienciaHipocalcemia | Frecuente: TF, tronco arterioso, CIV, CAI,Otros defectos asociados: TGV, CIA, TF, SCIH |
| Síndrome de Williams-Beuren | Deleción de 7q11.23 | Características dismórficas«Personalidad social», problemas psiquiátricosAnomalías endocrinasAnomalías óseas y del tejido conjuntivo | Frecuente: EASVOtros defectos asociados: CVSP, EPP |
| Síndrome de Jacobsen | Deleción de 11q23 terminal | Características dismórficasRetraso del crecimiento y discapacidad intelectualTrombocitopenia y disfunción plaquetariaImmunodeficiencia | Frecuente: CIV, anomalías de la válvula mitral, VAB, SCIHOtros defectos asociados: TGV, CAV, EVP, PDA |
| Síndrome de deleción de 1p36 | Deleción de 1p36 | Características dismórficasDiscapacidad intelectualAnomalías estructurales cerebralesDéficit visuales y auditivosObesidad | Frecuente: CIA, CIV, Ebstein, PDA, TFOtros defectos asociados: ACVI, MCD |
| Síndrome de deleción de 8p23.1 | Deleción de 8p23.1 | Hernia diafragmática | CIA, corazón triauricular, CIV, TF |
| Variación de un solo gen | |||
| Síndrome de Alagille | JAG-1, NOTCH-2 | Características faciales típicasColestasisEmbriotoxon posteriorVértebras en mariposa (somatosquisis) | Frecuente: EPP, EASVOtros defectos asociados: CIA, CIV, TF |
| Síndrome de Holt-Oram | TBX5 | Defectos del rayo radial | Frecuente: CIA (monoaurícula), anomalías de la conducción |
| Síndrome de Char | TFAP2B | Características faciales típicasAplasia/hipoplasia de falange media del quinto dedoDH leve | Frecuente: PDA |
| Síndrome de Ellis-Van Creveld | EVC* | Características faciales típicasBaja estatura rizomélica, polidactiliaHipoplasia unguealAnomalías dentalesFrenillo oral | Frecuente: CIA (monoaurícula)Otros defectos asociados: anomalías mitrales y tricuspídeas, PDA, CIV, SCIH |
| Síndrome de Adams-Oliver | ARHGAP31DOCK6*RBPJDLL4NOTCH1 (en su mayor parte asociado a CPC) | Defectos de la piel y el cuero cabelludoAnomalías de extremidades: sindactilia, polidactilia, falange distal cortaMicrocefalia, y trastorno del desarrollo en 1/3 | Frecuente: VM en paracaídas, VAB, EA, CoA, SCIHOtros defectos asociados: TF, CIA, CIV, tronco arteriosus |
| Síndrome de Kabuki | MLL2KDM6A | Características faciales típicasDiscapacidad intelectualAnomalías óseas: escoliosis, displasia de cadera, anomalías vertebralesAnomalías urogenitales | Frecuente: VAB, CoA, SCIH, CIA, CIVOtros defectos asociados: RVPPA, TF, EVP, anomalías de la válvula mitral |
| Síndrome CHARGE | CHD7 | ColobomaAtresia de coanaRetraso del crecimiento y discapacidad intelectualAnomalías urogenitalesAnomalías de la oreja y déficit auditivoParálisis de pares craneales | Frecuente: TF, CAI, tronco arteriosusOtros defectos asociados: anillos vasculares, CAV, CIV, PDA |
| Síndrome de Noonan | PTPN11 (asociado a EVP)SOS1 (asociado a CIA) RAF1 (asociado a MCH)Otros: RIT1, KRAS, SHOC2, NRAS, SOS2 y BRAF | Características faciales típicas, cuello aladoBaja estatura y retraso puberalHipotiroidismoAnomalías hematológicas y cánceresLinfedema | Frecuente: MCH, EVPOtros defectos asociados: defecto de la válvula mitral, CIA, CoA, TF |
| Síndrome de Costello | HRAS | Características faciales toscasPliegues palmoplantares profundosHipotonía, problemas de alimentaciónMayor riesgo de cáncerDiscapacidad intelectual | Frecuente: MCH, EVPOtros defectos asociados: defecto de la válvula mitral, CIA, CoA, TF |
| Síndrome de Leopard | PTPN11RAF1BRAF | Lentigos múltiples en cara, espalda y parte superior del troncoCaracterísticas faciales típicas | Frecuente: MCH, EVPOtros defectos asociados: defecto de la válvula mitral, CIA, CoA, TF |
| Síndrome de Myhre | Características faciales típicasDiscapacidad intelectual leveAutismoBaja estaturaPiel gruesaContracturas articularesCataratas | Estenosis aórtica, TF | |
ACVI: ausencia de compactación del ventrículo izquierdo; CAI: cayado aórtico interrumpido; CAV: comunicación auriculoventricular; CIA: comunicación interauricular; CIV: comunicación interventricular; CNV: variación en el número de copias; CoA: coartación de aorta; CPC: cardiopatías congénitas; EASV: estenosis aórtica supravalvular; EVP: estenosis de la válvula pulmonar; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; PDA: persistencia del ductus arteriosus; RVPA(T): retorno venoso pulmonar anormal (total); RVPPA: retorno venoso pulmonar parcialmente anormal; SCIH: síndrome de corazón izquierdo hipoplásico; TF: tetralogía de Fallot; TGV: transposición de grandes vasos; VAB: válvula aórtica bicúspide.
La identificación de genes en la CPC aislada se ha visto dificultada por varios factores: en primer lugar, defectos en genes diferentes pueden dar lugar a fenotipos similares, y diferentes fenotipos pueden ser consecuencia de defectos en el mismo gen. En segundo lugar, sobre todo en los casos esporádicos, la CPC puede tener una causa multifactorial. A pesar de las técnicas de detección sistemática molecular de gran capacidad, la identificación de la enfermedad multifactorial se encuentra aún en su primera infancia. Se han propuesto avances recientes en las puntuaciones de riesgo poligénicas para el cáncer familiar y las miocardiopatías. Es de esperar que suceda lo mismo en el caso de las CPC.
APLICACIONES PRÁCTICAS DE LA EVALUACIÓN GENÉTICA EN LAS CPCJunto con el aumento del conocimiento, estamos asistiendo a una ampliación continua de las repercusiones clínicas de las pruebas genéticas en las CPC. Sin embargo, para que las pruebas genéticas sean parte integrante de la asistencia estándar de los pacientes con CPC, hay varias consideraciones a las que se debe prestar atención.
En primer lugar, las pruebas genéticas deben tenerse en cuenta claramente en ciertos subgrupos de pacientes: los que presentan manifestaciones sindrómicas y los que tienen múltiples familiares afectados son los que muestran una mayor probabilidad de estar afectados por un problema genético subyacente. En segundo lugar, las pruebas genéticas y el asesoramiento genético deben personalizarse para cada paciente individual. La elección de la prueba genética, así como el momento adecuado para realizarla, deben decidirse de manera individualizada. En tercer lugar, cuando se identifica una causa genética de la CPC, ello debe acompañarse de un asesoramiento para comentar el tratamiento apropiado del paciente y su familia y, en los casos en los que esté indicado, el riesgo de recurrencia.
Por último, aunque desde luego no por ello menos importante, las pruebas genéticas deben tener en cuenta los aspectos psicológicos, sociales y culturales. La decisión de realizar estas pruebas debe tomarse de manera bien informada y de tal modo que sea el paciente quien tenga la última palabra.
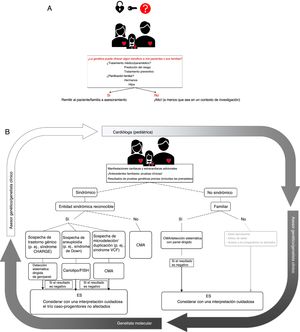
Algunas de estas cuestiones se comentan con más detalle a continuación y se ilustran en la figura 1.
, en colaboración con genetistas clínicos y asesores genéticos, verificarán la posible presencia de otras características clínicas cardiacas para descartar entidades sindrómicas. Basándose en ello, después se realizarán escalonadamente las pruebas genéticas apropiadas. La secuenciación de exoma es un paso final que debe considerarse e interpretarse cuidadosamente. Los resultados se enviarán al clínico y luego a los pacientes con el asesoramiento apropiado. CMA: análisis de microchip cromosómico; ES: secuenciación de exoma; FISH: hibridación in situ fluorescente; VCF: síndrome velocardiofacial.")
El proceso de evaluación genética en la cardiopatía congénita. A: sopesar cuidadosamente el posible beneficio de una evaluación genética. B: algoritmo para las pruebas clínicas/moleculares. En un primer paso, los cardiólogos (pediátricos), en colaboración con genetistas clínicos y asesores genéticos, verificarán la posible presencia de otras características clínicas cardiacas para descartar entidades sindrómicas. Basándose en ello, después se realizarán escalonadamente las pruebas genéticas apropiadas. La secuenciación de exoma es un paso final que debe considerarse e interpretarse cuidadosamente. Los resultados se enviarán al clínico y luego a los pacientes con el asesoramiento apropiado. CMA: análisis de microchip cromosómico; ES: secuenciación de exoma; FISH: hibridación in situ fluorescente; VCF: síndrome velocardiofacial.
La primera situación en la que se deben tener en cuenta las pruebas genéticas es la presencia de otras anomalías extracardiacas que indiquen una entidad sindrómica. Para identificar correctamente a los niños y adultos que forman parte de este grupo, es esencial un examen cuidadoso de la historia clínica y una caracterización fenotípica detallada por un genetista clínico. Además, puede ser preciso un examen de detección sistemática fenotípica de los familiares de primer grado para identificar las posibles manifestaciones sindrómicas. Las manifestaciones clínicas que deben levantar la sospecha de un problema sindrómico subyacente son la discapacidad intelectual o los déficit sensitivos, la presencia de características dismórficas o la baja estatura, así como la concomitancia de otros trastornos congénitos o endocrinos15,60.
La segunda situación en la que se debe considerar la posible conveniencia de una evaluación genética es la que se da cuando hay múltiples familiares afectados. Las formas familiares de CPC constituyen una parte pequeña del total de las CPC, tal como se refleja en un estudio de población danesa, en el que solo un 2,2% de las CPC eran de tipo familiar4. No obstante, puede alcanzar un alto rendimiento diagnóstico en algunas familias, como indican los casos familiares de estenosis aórtica supravalvular, en los que puede hallarse una afección genética en el 85% de las familias61.
También se puede considerar hacer pruebas genéticas a recién nacidos y lactantes con CPC. Las características genéticas son importantes factores que determinan alteraciones del neurodesarrollo y lesiones extracardiacas en los niños con CPC62,63. El conocimiento de esta predisposición genética podría mejorar los resultados a largo plazo en esos niños.
Además de las indicaciones mencionadas, es preferible remitir a todo paciente adulto con una CPC y el deseo activo de tener hijos (tanto varones como mujeres) a un asesoramiento genético y la posible realización de pruebas. La aparición de nuevas técnicas preconcepcionales y prenatales permite evitar la transmisión de la CPC a la siguiente generación. Si hay un trastorno genético subyacente desconocido, el asesoramiento genético continúa siendo muy importante para estimar el riesgo de recurrencia.
Como ya se ha mencionado, en los pacientes adultos con una CPC que ya han pasado una evaluación genética con técnicas más antiguas, podrían aportar beneficio una nueva evaluación y la repetición de las pruebas.
Individualización de las pruebas genéticasLa elección de la prueba genética más apropiada para cada paciente individual con CPC es muy importante y requiere una colaboración estrecha entre los genetistas clínicos y moleculares. Se ha observado que un examen cuidadoso previo a la prueba por un asesor genético puede reducir la proporción de pruebas inadecuadas en un 26%64. La elección de la técnica durante el diagnóstico genético depende en gran medida de la forma de presentación clínica y de los antecedentes familiares. En la figura 1 se muestra un diagrama de flujo que indica el modo de ajustar individualmente esas técnicas a un paciente concreto.
Acciones motivadas por los resultados genéticosAl remitir a un paciente a asesoramiento genético y pruebas genéticas, la cuestión clave es siempre si la identificación de un defecto genético puede aportar un beneficio al paciente o a su familia. A este respecto, es importante tener en cuenta estos argumentos:
Mejora del tratamientoEl conocimiento de un problema genético subyacente puede ser importante para diagnosticar y mejorar el tratamiento de las complicaciones extracardiacas de los niños y los adultos con CPC. Por ejemplo, los pacientes con una deleción de 22q11 pueden presentar una disminución de la inmunidad de linfocitos T y, por consiguiente, pueden tener un riesgo de enfermedades infecciosas graves; los pacientes con síndrome de Alagille pueden sufrir complicaciones oftálmicas y hepáticas; los niños con síndrome de Noonan pueden tener estatura baja, por lo que es posible que les aporte beneficio un tratamiento con hormona del crecimiento.
Algunos defectos genéticos se han asociado con un aumento del riesgo de otras complicaciones cardiovasculares. Un ejemplo conocido es la asociación entre la CIA y los trastornos de la conducción en los pacientes portadores de una variante patógena en el gen NKX2.5. En estos pacientes son más probables el bloqueo auriculoventricular, la disfunción ventricular y la muerte súbita cardiaca65. Diversos tipos de CPC se han asociado con genes causantes de una miocardiopatía familiar. Algunos ejemplos son los genes ACTC1, MYH6 y MYBPC366-68.
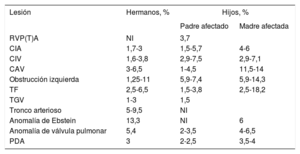
Riesgo de recurrencia y consecuencias para otros familiaresAl comentar el riesgo de recurrencia, el conocimiento de la entidad genética subyacente resulta esencial. Las estimaciones del riesgo variarán también en función del contexto de hermanos o hijos, y en algunos casos los riesgos son diferentes para el padre y la madre. Si hay un trastorno genético conocido, el riesgo de recurrencia en un hermano dependerá en gran parte del tipo de herencia y de si la anomalía es de nueva aparición o no. En los trastornos genéticos que tienen un patrón autosómico recesivo, el riesgo de recidiva es de un 25%. En los que tienen un patrón autosómico dominante, el riesgo de recurrencia será del 50% si está afectado uno de los progenitores y de hasta un 1% en el caso de que sea una variación de novo69. En los pacientes adultos con una CPC que tienen un trastorno genético conocido, el riesgo de recurrencia en los hijos será del 50% si el trastorno es autosómico dominante. En los pacientes con anomalías de herencia autosómica recesiva, el riesgo de recidiva en los hijos es similar al de la población general, pero cada hijo será portador de 1 alelo con la anomalía. Si no se encuentra una anomalía genética subyacente, el riesgo de recurrencia continúa siendo superior al de la población general. Si se considera el conjunto de todas las CPC, el riesgo de recurrencia en los hermanos se estima en un 2,1% y el de recurrencia en los hijos, en un 4,4%; y en general las mujeres tienen una tasa de recurrencias superior a la de los varones70. En la tabla 4 se resume el riesgo de recurrencia epidemiológico conocido de las CPC más frecuentes en ausencia de una causa genética subyacente conocida. Algunas lesiones como la heterotaxia, la obstrucción del tracto de salida del ventrículo derecho y la comunicación auriculoventricular muestran una mayor agrupación familiar. El riesgo de recurrencia es de 80 a 25 veces superior al de la población general4.
Riesgo de recurrencia de la CPC en hermanos e hijos de pacientes con una CPC no sindrómica y sin un defecto molecular conocido5,70-74
| Lesión | Hermanos, % | Hijos, % | |
|---|---|---|---|
| Padre afectado | Madre afectada | ||
| RVP(T)A | NI | 3,7 | |
| CIA | 1,7-3 | 1,5-5,7 | 4-6 |
| CIV | 1,6-3,8 | 2,9-7,5 | 2,9-7,1 |
| CAV | 3-6,5 | 1-4,5 | 11,5-14 |
| Obstrucción izquierda | 1,25-11 | 5,9-7,4 | 5,9-14,3 |
| TF | 2,5-6,5 | 1,5-3,8 | 2,5-18,2 |
| TGV | 1-3 | 1,5 | |
| Tronco arterioso | 5-9,5 | NI | |
| Anomalía de Ebstein | 13,3 | NI | 6 |
| Anomalía de válvula pulmonar | 5,4 | 2-3,5 | 4-6,5 |
| PDA | 3 | 2-2,5 | 3,5-4 |
CAV: comunicación interventricular; CIA: comunicación interauricular; CIV: comunicación interventricular; CPC: cardiopatías congénitas; NI: no indicado; PDA: persistencia del ductus arteriosus; RVPA(T): retorno venoso pulmonar anormal (total); TF: tetralogía de Fallot; TGV: transposición de grandes vasos.
Otro aspecto importante por lo que respecta al asesoramiento de los familiares es la necesidad de una evaluación clínica adicional. Para algunas lesiones, como la obstrucción del TSVI, hay diferencias conocidas en el espectro clínico que van de una VAB asintomática a un síndrome del hemicardio izquierdo hipoplásico grave75. En un estudio, el riesgo relativo de que un progenitor o un hermano de un paciente con una lesión obstructiva del TSVI tuviera una VAB fue 5,05 (intervalo de confianza del 95%, 2,2-11,7)11. Esta gran incidencia, junto con el hecho de que muchas de las complicaciones de las lesiones del lado izquierdo son tratables o prevenibles, llevó a justificar la conveniencia de realizar una evaluación ecocardiográfica de los familiares de primer grado de los pacientes con obstrucción del TSVI76. Actualmente no se recomienda un examen de detección sistemático en los familiares para otras formas de CPC no sindrómicas.
Diagnóstico prenatal y examen de detección sistemática fetalEl diagnóstico prenatal es posible siempre que se haya identificado una causa genética de la CPC. En este caso, puede impedirse la transmisión a la siguiente generación mediante el diagnóstico preimplantacional, que es una técnica basada en la fecundación in vitro, que se aplica antes del embarazo y permite seleccionar los embriones no afectados.
Una alternativa son las pruebas prenatales convencionales, que en general implican la obtención de muestras de vellosidades coriónicas o líquido amniótico en las fases iniciales del embarazo. En los últimos años han aparecido las pruebas prenatales no invasivas (NIPT) como alternativa no invasiva a las pruebas prenatales. Las NIPT se desarrollaron principalmente para detectar la trisomía 21 en el feto en una fase temprana del embarazo con altas especificidad (> 99%) y sensibilidad (> 99%) en ausencia de anomalías fetales. Durante el embarazo, hay células de la placenta (que contienen ADN fetal) que sufren una lisis y liberan su contenido a la circulación materna. La prueba se basa en el número relativo de lecturas asignadas en el mapeo a un determinado cromosoma en el plasma materno (ADN acelular). Así pues, la prueba permite detectar otras aneuploidías como la 13 y 18, el síndrome de Klinefelter (47,XXY), el síndrome de Turner (45,X0) o el síndrome triple X (47,XXX), aunque con sensibilidad y especificidad algo inferiores. En el futuro, un ajuste fino de la tecnología acabará haciendo que las NIPT sean apropiadas para detectar variantes de nueva aparición (tanto CNV como SNV)77,78 y para realizar pruebas dirigidas de variantes heredadas79. Ni que decir tiene que la aplicación a gran escala de las NIPT requiere una consideración cuidadosa de las cuestiones éticas. En ambos casos de pruebas prenatales, puede considerarse la posibilidad de interrumpir el embarazo si los resultados de la prueba son anormales.
Algunas parejas pueden optar por no realizar el diagnóstico prenatal, en cuyo caso debe comentarse la posibilidad de un examen genético de detección sistemática en el recién nacido. Si no se ha identificado una causa genética subyacente de la CPC o no se han realizado pruebas prenatales, se recomienda una ecocardiografía fetal. Esta debe realizarse en un centro especializado a las 18-20 semanas de embarazo80.
CONCLUSIONESLa evaluación genética de los pacientes con una CPC se está realizando a escala cada vez mayor y es indudable que, en muchos casos, ayuda a optimizar el tratamiento (para)médico de pacientes individuales y sus familias. Además, el conocimiento de la genética también ayuda a comprender la fisiopatología subyacente a estos trastornos, y ello contribuirá ciertamente a desarrollar, a largo plazo, más tratamientos dirigidos.
La evaluación genética debe realizarse correctamente en cada paciente/familia con un asesoramiento cuidadoso antes de practicar las pruebas genéticas, así como para la comunicación de cualquier resultado obtenido. Está claramente demostrado que la aplicación de una estrategia correcta conduce a unas pruebas más eficientes, mayor satisfacción del paciente y un tratamiento médico más correcto.
FINANCIACIÓNJ. De Backer y B. Callewaert han contado con financiación como investigadores clínicos sénior de la Flanders Research Foundation. J. De Backer ha recibido subvención para investigación médica del Baillet Latour Fund.
CONFLICTO DE INTERESESLos autores no tienen conflictos de intereses.