En este artículo de revisión se comentan las bases genéticas de la parada cardiaca, prestando especial atención a las canalopatías cardiacas y la miocardiopatía ventricular derecha. Revisamos el uso apropiado de las pruebas genéticas para pacientes en quienes se sospecha de arritmias cardiacas hereditarias, subrayando la importancia de la mayoría de las correlaciones genotipo-fenotipo para la estratificación del riesgo. El artículo presenta también las opiniones más recientes sobre los criterios diagnósticos y los diagramas de flujo para el tratamiento de los pacientes con enfermedades arritmogénicas hereditarias.
Palabras clave
En las últimas dos décadas, se ha descrito un número creciente de enfermedades arritmogénicas hereditarias, y se han atribuido varios casos de arritmias inexplicadas en individuos jóvenes a trastornos heredables bien definidos. La identificación de mutaciones en los genes que causan estas enfermedades ha facilitado un conocimiento progresivo de su fisiopatología1, 2 y ha proporcionado al clínico nuevos instrumentos para la estratificación del riesgo y el tratamiento basados en la genética3. El análisis genético ha pasado a ser un instrumento importante para identificar el sustrato molecular en los pacientes afectados por una enfermedad arritmogénica hereditaria o en los que se sospecha su presencia.
A pesar de la heterogeneidad de los sustratos y de la expresividad clínica, las pruebas genéticas tienen repercusiones directas en la práctica clínica: permiten al médico establecer/perfeccionar el diagnóstico incluso en portadores silentes, y en algunas enfermedades la identificación de una mutación tiene consecuencias importantes para la estratificación del riesgo y el tratamiento de los pacientes3. En la presentación general que sigue se aborda el papel de las pruebas genéticas para cada una de las enfermedades arritmogénicas hereditarias epidemiológicamente relevantes: síndrome de QT largo (SQTL), síndrome de Brugada (SBr), síndrome de QT corto (SQTC), taquicardia ventricular polimórfica catecolaminérgica (TVPC) y miocardiopatía ventricular derecha arritmogénica (MVDA).
Síndrome de qt largoEl SQTL se caracteriza por una prolongación excesiva de la repolarización ventricular y un aumento del riesgo de taquiarritmias ventriculares malignas en pacientes con un corazón morfológicamente indemne4. La prevalencia estimada es de entre 1:2.500 y 1:5.000. Sin embargo, dado que probablemente hasta dos tercios de los pacientes no están identificados y que un 10-35% tiene un intervalo QT corregido (QTc) normal, es probable que la prevalencia real sea más alta5, 6. La media de edad de inicio de los síntomas (síncope o muerte súbita) es de 12 años y un inicio más temprano se asocia generalmente a una evolución más grave7.
El síncope (debido a una taquicardia ventricular [TV] polimórfica autolimitada) y la parada cardiaca, a menudo desencadenada por una activación adrenérgica aguda, son las manifestaciones características. Cuando se ha establecido el diagnóstico, se hace posible la prevención de los episodios arrítmicos con peligro para la vida. El tratamiento antiadrenérgico (bloqueadores beta) y el empleo de desfibrilador automático implantable (DAI) constituyen la esencia del tratamiento del SQTL. El uso apropiado de este arsenal terapéutico u otros tratamientos adicionales, como las terapias génicas específicas y la simpatectomía izquierda en casos seleccionados, con frecuencia resulta eficaz para reducir el riesgo de muerte. En el electrocardiograma (ECG) de superficie, se puede observar una duración del intervalo QTc que supera los valores normales (es decir, intervalos QT > 440 ms en los varones y > 460 ms en las mujeres)8.
Análisis genético en el síndrome de QT largoEn la mayoría de los casos, el SQTL se transmite como una enfermedad autosómica dominante, el síndrome de Romano-Ward. La forma autosómica recesiva, denominada síndrome de Jervell y Lange-Nielsen, se caracteriza por la coexistencia de prolongación del QT y sordera congénita.
Los primeros tres genes del SQTL identificados por el grupo de Keating fueron el KCNQ1, que codifica la proteína que facilita la corriente de potasio IKs; el KCNH2, que codifica el canal de la corriente de repolarización de potasio IKr, y el SCN5A, que codifica la subunidad a del canal de sodio que facilita la corriente de despolarización de sodio INa8.
En los últimos 15 años, se han descubierto en los pacientes con SQTL mutaciones en otros genes que codifican subunidades de diversos canales iónicos, así como mutaciones en genes que codifican proteínas reguladoras de los canales iónicos. Hasta el momento se han publicado 13 variantes diferentes del SQTL (Tabla 1).
Tabla 1. Genes asociados al síndrome de QT largo
| Fenotipo | Variante | Gen | Proteína | Defecto funcional |
| Síndrome de QT largo | LQT1 | KCNQ1 | KvLQT1 (subunidad alfa del canal de potasio) | Pérdida de función |
| LQT2 | KCNH2 | HERG (subunidad alfa del canal de potasio) | Pérdida de función | |
| LQT3 | SCN5A | Nav1.5 (subunidad alfa del canal de sodio) | Ganancia de función | |
| LQT4 | ANK2 | Ankirina B, proteína de anclaje | Pérdida de función | |
| LQT5 | KCNE1 | MinK (subunidad beta del canal de potasio) | Pérdida de función | |
| LQT6 | KCNE2 | MiRP (subunidad beta del canal de potasio) | Pérdida de función | |
| LQT7, síndrome de Andersen | KCNJ2 | Kir2.1 (subunidad alfa del canal de potasio) | Pérdida de función | |
| LQT8, síndrome de Timothy | CACNA1c | Cav1.2 (subunidad alfa del canal de calcio tipo L) | Ganancia de función | |
| LQT9 | CAV3 | Gen de caveolina cardiaca | Ganancia de función | |
| LQT10 | SCN4B | Subunidad β4 del canal de sodio | Ganancia de función | |
| LQT11 | AKAP9 | Proteína de anclaje de cinasa A | Reducción de la corriente IKs | |
| LQT12 | SNTA1 | Sintrofina | Aumento de corriente de sodio | |
| LQT13 | KCNJ5 | Subunidad Kir 3.4 de canal IKAch | Pérdida de función |
Los genes del SQTL afectan a las corrientes iónicas, bien directamente (mutaciones de canales iónicos), bien indirectamente (chaperonas u otros moduladores)9. Dos de las variantes, LQT7 y LQT8, se caracterizan por la presencia de un fenotipo extracardiaco que da lugar a dos síndromes claramente diferenciados. La LQT7 (síndrome de Andersen), causada por mutaciones en el gen KCNJ2, es una variante muy poco frecuente (< 1%) e incluye parálisis periódica y manifestaciones dismórficas. El ECG se caracteriza por la presencia de ondas U marcadas y arritmias ventriculares como la TV bidireccional10. La LQT8 (síndrome de Timothy)11 está causada por mutaciones de ganancia de función en el gen CACNA1c que codifica el canal del calcio dependiente de voltaje de tipo L. Es interesante señalar que más del 90% de los pacientes afectados por la variante LQT8 son portadores de las mismas mutaciones G406R; esto difiere de lo que se observa en la otra variante genética del SQTL, que se caracteriza por la extrema heterogeneidad de diferentes mutaciones. Los pacientes con síndrome de Timothy presentan una prolongación marcada del intervalo QT asociada a un fenotipo complejo que incluye sindactilia, bloqueo auriculoventricular, cardiopatías congénitas, autismo, trastornos del desarrollo y reducción de la respuesta inmunitaria.
Se identificó un defecto genético en un 60-72% de los pacientes con SQTL12, y más del 90% de los pacientes en quienes se ha determinado el genotipo corresponden a las primeras tres variantes (LQT1, LQT2 y LQT3). Por lo tanto, actualmente no está indicado el examen de detección aplicado de forma amplia. El análisis de los signos del ECG, junto con los antecedentes personales y familiares, puede ser útil para orientar el enfoque de uso de la prueba13. Es de destacar que la probabilidad de un genotipo positivo es máxima (hasta un 72%) cuando la determinación se realiza en individuos con la máxima probabilidad fenotípica de tener el síndrome14, lo que refuerza el papel de la correlación fenotípica para orientar las decisiones racionales en cuanto a las determinaciones del genotipo.
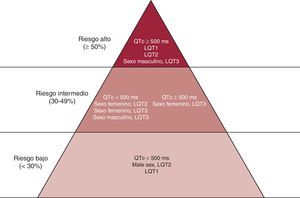
El examen de detección de ANK2, KCNJ2 y CACNA1c sólo está indicado en presencia de fenotipos específicos que apunten a la alteración de esos genes. El genotipo tiene una repercusión clínica directa en el SQTL: se han descrito diferencias específicas según el gen en cuanto a la morfología de los complejos de ondas ST-T, los desencadenantes de los episodios cardiacos15 y el riesgo de estos episodios7, 16, 17. Los estudios de genotipo-fenotipo han aportado información importante sobre el efecto de la localización, el tipo de codificación y la función biofísica de las mutaciones de los canales sobre las manifestaciones fenotípicas y el curso clínico de los pacientes con SQTL18. Según estas observaciones, hay consenso respecto a que los genotipos LQT2 y LQT3 muestran peor pronóstico y una respuesta relativamente mala al tratamiento con bloqueadores beta. Por lo que respecta al tratamiento de los pacientes en quienes se ha determinado el genotipo, el tratamiento con bloqueadores beta se ha asociado a una reducción significativa de la tasa de episodios cardiacos en los pacientes con mutaciones de LQT1 y LQT2, pero no ha habido ninguna reducción evidente en los que presentan mutaciones de LQT319. Se puede considerar la implantación de un DAI para la prevención primaria de la parada cardiaca súbita en pacientes con estas variantes del SQTL cuando se asocian a un QTc > 500 ms e inicio temprano de episodios cardiacos (edad < 7 años). Podemos concluir que el locus de la mutación causal afecta al curso clínico del SQTL y, junto con el sexo, modula los efectos del QTc en las manifestaciones clínicas. Partiendo de estos datos, nuestro grupo propuso un enfoque para la estratificación del riesgo basada en estas variables (Figura 1).

Figura 1. Estratificación del riesgo en el síndrome de QT largo, según la duración del intervalo QTc, el genotipo y el sexo. QTc: QT corregido. Reproducido con permiso de Priori et al 7 .
En esta breve revisión es fácil observar que la determinación del genotipo puede tener una repercusión clínica importante en el plan de asistencia de los pacientes con SQTL y, de hecho, está en disposición de pasar ya a un primer plano.
Influencia del genotipo en el tratamiento clínico del síndrome de QT largoAunque la heterogeneidad genética en el SQTL es considerable, hay tres genes que intervienen de manera principal y explican más del 90% de los casos de SQTL: KCNQ1 (LQT1), KCNH2 (LQT2) y SCN5A (LQT3). Estas tres variantes genéticas del SQTL de mayor prevalencia presentan diferencias en sus manifestaciones, indicadores de riesgo y respuesta al tratamiento. Gracias a estos descubrimientos, el genotipo ha pasado a formar parte del esquema de estratificación del riesgo de los pacientes con SQTL.
Las primeras correlaciones genotipo-fenotipo observadas en el SQTL apuntaban a que los pacientes con LQT1 presentan la mayor parte de los síntomas durante las actividades deportivas, en especial la natación, mientras que los individuos con LQT3 tienen mayor riesgo de arritmias durante el sueño o en reposo y los pacientes con LQT2 sufren episodios asociados al ruido intenso15. Se puso claramente de manifiesto que, junto con el sexo y la duración del intervalo QT, el genotipo es un factor determinante del riesgo de muerte súbita cardiaca (MSC) y la respuesta al tratamiento7, 16. Tal como se muestra en la Figura 1, las mujeres con LQT2 y los varones con LQT3 que presentan un intervalo QT > 500 ms se encuentran en la categoría de mayor riesgo, con independencia de otros factores7. De manera análoga, mientras que los pacientes con LQT1 tienden a presentar una respuesta óptima a los bloqueadores beta, los pacientes con LQT2 o LQT3 sufren recurrencias a pesar de emplearlos a dosis plenas16. Basándose en estos datos y en la opinión de expertos, las guías del American College of Cardiology/American Heart Association/Sociedad Europea de Cardiología 2006 para la prevención de la MSC20 indican que la presencia de un intervalo QTc > 500 ms en LQT2 y LQT3 abre la posibilidad de implantación de un DAI profiláctico (Figura 1). Es interesante que, partiendo de la evidencia de que la variante LQT3 está causada por un exceso de la corriente de entrada de sodio, se propusiera que la mexiletina podía ejercer un efecto antiarrítmico en los pacientes con LQT3 al reducir la duración del intervalo QTc a través de un efecto de bloqueo del canal de sodio21.
Recientemente se ha obtenido evidencia clínica de que la respuesta clínica a la mexiletina se ve influida por el tipo de mutación y que a menudo la mexiletina podría ser ineficaz22 o incluso nociva23.
Síndrome de BrugadaEl SBr es una enfermedad arritmogénica hereditaria, caracterizada por un patrón electrocardiográfico específico, con elevación del segmento ST en las derivaciones V1 a V3 y un bloqueo de rama derecha del haz, completo o incompleto, en ausencia de cardiopatía estructural24.
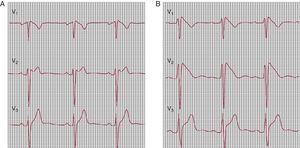
Sólo un patrón concreto del segmento ST se considera diagnóstico: la elevación en V1, V2 y V3 con morfología convexa (coved) de al menos 2 mm, el denominado ECG de tipo I (Figura 2B). Este patrón diagnóstico puede ser intermitente. Con bastante frecuencia, el ECG de presentación indica un posible SBr (elevación del segmento ST de tipo «silla de montar» [saddle-back]) (Figura 2A). La administración intravenosa de un solo bolo de un bloqueador del canal del sodio (procainamida, flecainida o ajmalina) puede ocasionar la conversión de un patrón oculto o no diagnóstico a un patrón de tipo I coved25, 26 (Figura 2).

Figura 2. Prueba de flecainida en un paciente en el que se sospecha un síndrome de Brugada. A: electrocardiograma basal (tipo 2). B: flecainida 2mg/kg; patrón diagnóstico (tipo 1).
La prevalencia mundial estimada es de un 0,10%27, y es posible que sea superior en áreas endémicas del sudeste asiático28. La mayoría de los pacientes con diagnóstico clínico son varones, aunque todavía no se conoce cómo el sexo modula la manifestación de la enfermedad24, 25, 29.
Las manifestaciones clínicas más frecuentes del SBr son el síncope o la MSC causados por taquiarritmias ventriculares que se producen sobre todo durante el sueño o en reposo; otros desencadenantes arrítmicos podrían ser la fiebre o las comidas abundantes. También hay arritmias supraventriculares como la fibrilación auricular en un 15-20% de los pacientes con diagnóstico de SBr30. El bloqueo auriculoventricular y los retrasos de la conducción intraventricular forman parte del fenotipo del SBr31. Por extensión, la presencia de potenciales tardíos debe considerarse un indicador clínico de la enfermedad, puesto que se dan en un 50% de los pacientes con afección clínica.
Hasta el momento ningún tratamiento médico ha resultado eficaz para prevenir las arritmias y la muerte súbita en el SBr; la implantación de un DAI es el único tratamiento disponible32. La infusión de isoproterenol es útil para el control agudo de las tormentas arrítmicas. El único fármaco que se ha utilizado hasta ahora con cierto éxito es la quinidina33; su uso se puede considerar para el tratamiento adyuvante en casos de arritmias recurrentes y descargas frecuentes del DAI. Lamentablemente, el tratamiento a largo plazo con quinidina se asocia a un porcentaje de abandonos importante a causa de sus efectos secundarios gastrointestinales.
Por lo tanto, es crucial la estratificación del riesgo para seleccionar a los pacientes de alto riesgo en quienes es probable que se obtenga un beneficio con un DAI. Hay acuerdo general respecto a que los antecedentes de síncope en presencia de un patrón de ECG de tipo I identifican a los pacientes con mayor riesgo de MSC32. Cuando mediante provocación farmacológica se produce un ECG de tipo I que nunca se produce espontáneamente, el riesgo arrítmico es inferior32.
Lamentablemente, un porcentaje elevado de los pacientes con diagnóstico de SBr se encuentran en la categoría de riesgo intermedio, puesto que están asintomáticos y pueden presentar un patrón de tipo I de forma espontánea. En consecuencia, los clínicos y los científicos se enfrentan al reto de identificar nuevos marcadores que permitan estratificar mejor el riesgo arrítmico en ese grupo y así seleccionar a los pacientes en los que debe implantarse un DAI. Aunque se propuso la estimulación eléctrica programada como medida para la estratificación del riesgo, se están acumulando evidencias en contra de su utilidad clínica32, 34, 35, 36; los antecedentes familiares de muerte súbita o la presencia de una mutación genética no influyen en el riesgo arrítmico. Datos recientes indican que la presencia de una fragmentación del QRS puede estar correlacionada con peor pronóstico36, 37.
Análisis genético en el síndrome de BrugadaLa enfermedad se transmite en forma de rasgo autosómico dominante. El componente genético del síndrome se atribuye a las mutaciones de 10 genes diferentes (Tabla 2). El primer gen relacionado con el SBr se descubrió en 1998 y es el SCN5A, que codifica el canal del sodio cardiaco; se trata del mismo gen que está ligado al LQT3. A día de hoy, la mayor parte de los pacientes en quienes se ha determinado el genotipo son portadores de una mutación en ese gen38.
Tabla 2. Genes asociados a enfermedades arritmogénicas hereditarias
| Fenotipo | Variante | Gen | Proteína | Defecto funcional |
| Síndrome de Brugada | BrS1 | SCN5A | Subunidad alfa del canal de sodio cardiaco (Nav1.5) | Pérdida de función |
| BrS2 | GPD1-L | Glicerol-6-fosfato-deshidrogenasa | Pérdida de función | |
| BrS3 | CACNA1c | Subunidad alfa del canal de calcio tipo L (Cav1.2) | Pérdida de función | |
| BrS4 | CACNB2 | Subunidad β2 del canal de calcio tipo L | Pérdida de función | |
| BrS5 | SCN1B | Subunidad β1 del canal de sodio cardiaco | Pérdida de función | |
| BrS6 | KCNE3 | Subunidad beta de corriente de salida transitoria | Ganancia de función | |
| BrS7 | SCN3B | Subunidad β3 del canal de sodio cardiaco | Pérdida de función | |
| BrS8 | MOG1 | Transporte nucleocitoplásmico y microtubular | Pérdida de función | |
| BrS9 | KCNE5 | Subunidad beta de corriente de salida transitoria | Ganancia de función | |
| BrS10 | KCND3 | Canal de potasio ITo (Kv4.3) | Ganancia de función | |
| Síndrome de QT corto | SQTS1 | KCNH2 | Subunidad alfa del canal de potasio IKr (HERG) | Ganancia de función |
| SQTS2 | KCNQ1 | Subunidad alfa del canal de potasio IKs (KvLQT1) | Ganancia de función | |
| SQTS3 | KCNJ2 | Canal de potasio IK1 (Kir2.1) | Ganancia de función | |
| SQTS4 | CACNA1c | Subunidad alfa del canal de calcio tipo L (Cav1.2) | Pérdida de función | |
| SQTS5 | CACNB2 | Subunidad β2 del canal de calcio tipo L | Pérdida de función | |
| SQTS6 | CACNA2D1 | Subunidad δ1 de canal de calcio tipo L | Pérdida de función | |
| TV catecolaminérgica | CPVT1 | RyR2 | Receptor de rianodina cardiaca (RyR2) | Liberación diastólica de calcio |
| CPVT2 | CASQ2 | Calsecuestrina cardiaca (CASQ2) | Liberación diastólica de calcio |
TV: taquicardia ventricular.
Está claro que la corriente de sodio desempeña un papel importante en la patogenia del SBr, tal como indica el hecho de que otros tres genes involucrados en la enfermedad (GPD1-L, SCN1B y SCN3B) influyan en la corriente INa39, 40, 41.
Recientemente, se ha relacionado con el fenotipo del SBr a los genes CACNA1c y CACNB2, que codifican las subunidades alfa y beta del canal del calcio cardiaco. Las mutaciones de pérdida de función de estos genes se han relacionado con la enfermedad42. Los primeros pacientes con SBr descritos en la literatura como portadores de mutaciones de los genes CACNA1c y CACNB2 presentaban un fenotipo bien definido que combinaba un intervalo QT corto con un patrón de ECG de SBr tipo I. Así pues, es importante evaluar con exactitud la presencia de un intervalo QT corto y/o un patrón de ECG de SBr como parte de un único fenotipo, con objeto de abordar mejor el empleo de pruebas de detección genéticas en esos pacientes.
Dado que no se conoce la prevalencia comparativa de mutaciones en los demás genes relacionados con el SBr (y es probable que sea muy baja), el examen de detección sistemática de estos genes, con la excepción del SCN5A, tiene una utilidad diagnóstica incierta. Además, es probable que la heterogeneidad genética del SBr sea aún mayor, ya que la detección sistemática de mutaciones en los genes conocidos permite identificar una mutación en el 25-30% de los pacientes clínicamente afectados. En consecuencia, el examen genético de detección sistemática es útil para confirmar el diagnóstico clínico y permite la identificación de portadores génicos silentes, pero a diferencia de lo que ocurre en el SQTL, hasta el momento no hay evidencia de que los resultados de las pruebas genéticas influyan en el tratamiento clínico o la estratificación del riesgo en el SBr32.
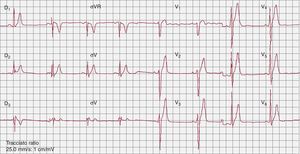
Síndrome de QT cortoEn 2000, Gussak et al43 identificaron una nueva enfermedad arritmogénica, caracterizada por un intervalo QT más corto de lo normal (< 350 ms) (Figura 3), arritmias ventriculares y auriculares y MSC. Teniendo en cuenta que sólo se ha descrito un pequeño número de pacientes con SQTC, parece que la MSC como primera manifestación no es infrecuente. Los intentos preliminares de estratificación del riesgo no han tenido éxito44 debido al bajo número de pacientes, y la implantación de un DAI es el tratamiento principal. Al prolongar el intervalo QT, el tratamiento con hidroquinidina parece ser eficaz para prevenir la inducción de la taquiarritmia ventricular y los episodios arrítmicos durante el seguimiento a largo plazo45.

Figura 3. Electrocardiograma basal en un paciente con síndrome de QT corto (QT/QTc, 300/300 ms).
Se han observado mutaciones en los genes KCNH2, KCNQ1, KCNJ2, CACNA1c y CACNB2 en individuos con diagnóstico de SQTC42, 46, 47, 48. Sin embargo, actualmente sólo se realiza la caracterización genética en una minoría de los pacientes.
Según datos recientes49, en aproximadamente un 20% de los pacientes con QT corto se determina con éxito el genotipo; por consiguiente, el valor de las pruebas genéticas en este síndrome es limitado y su uso no tiene consecuencias pronósticas. Los datos obtenidos en una pequeña cohorte de pacientes indican que los portadores de mutaciones del gen KCNH2 pueden presentar un intervalo QT más corto49. La estratificación del riesgo y el tratamiento del SQTC aún están mal definidos.
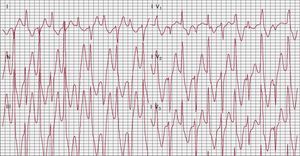
Taquicardia ventricular polimórfica catecolaminérgicaLa taquicardia ventricular polimórfica catecolaminérgica (TVPC) es un trastorno del manejo intracelular del calcio50. La prevalencia estimada es de 1:7.000-1:10.000. Es una de las enfermedades arritmogénicas hereditarias más letales, con una evolución natural en la que hasta un 30% de los pacientes sufren una MSC antes de los 40 años si no se emplea tratamiento antiadrenérgico51, 52. La enfermedad se manifiesta por arritmias de mecanismo adrenérgico, con peligro para la vida pues causan síncope o parada cardiaca, que se inician en la edad pediátrica. El ECG de superficie es anodino; en consecuencia, el diagnóstico se basa principalmente en los síntomas y la detección de arritmias inducidas por el estrés durante una prueba de esfuerzo o un registro Holter. Los pacientes presentan también arritmias supraventriculares, principalmente con salvas de taquicardia supraventricular o de fibrilación auricular que se solapan con extrasístoles ventriculares y TV. Durante la prueba de esfuerzo, es muy frecuente que los pacientes presenten un aumento progresivo de la complejidad de la arritmia hasta la aparición de una TV. La TV bidireccional es casi diagnóstica de la enfermedad: se caracteriza por una rotación de los complejos QRS en 180° de un latido a otro (Figura 4). Algunos pacientes no presentan TV bidireccional y durante el ejercicio sufren TV polimórfica51, 52.

Figura 4. Taquicardia ventricular bidireccional durante una prueba de esfuerzo en un paciente con taquicardia ventricular polimórfica catecolaminérgica.
Siempre que se establece el diagnóstico de TVPC, se debe administrar bloqueadores beta20. Aunque esto aporta protección a la mayoría de los pacientes, aproximadamente un 30% sufre al menos un episodio arrítmico durante el tratamiento51, 53, 54. Más recientemente, Watanabe et al55 han descrito la supresión de las arritmias en 2 pacientes con TVPC al tratarlos con flecainida oral. Aun cuando son necesarios datos de series más amplias antes de proponer el empleo de la flecainida como tratamiento médico en la TVPC, estos resultados son alentadores y justifican un examen más detallado.
Análisis genético en la taquicardia ventricular polimórfica catecolaminérgicaLos estudios realizados para identificar la base molecular de la TVPC llevaron a identificar en los individuos afectados mutaciones en dos genes que codifican proteínas del retículo sarcoplásmico: el receptor de rianodina (RyR2) y la calsecuestrina cardiaca (CASQ2), asociadas respectivamente a una forma autosómica dominante y una forma autosómica recesiva de TVPC56, 57.
Se ha demostrado que las mutaciones de los genes RyR2 y CASQ2 dan lugar a un aumento de la liberación de calcio procedente del retículo sarcoplásmico y fomentan la aparición de arritmias desencadenadas.
Alrededor del 70% de los pacientes en quienes se determina el genotipo son portadores de una mutación en el gen RyR251, mientras que la prevalencia de las mutaciones del CASQ2 es baja (aproximadamente un 7% en nuestra cohorte)58. El análisis genético tiene complicaciones logísticas derivadas del hecho de que el RyR2 es uno de los genes más grandes del genoma humano y el tiempo necesario para la determinación del genotipo es largo.
Un método coherente de detección de RyR2/CASQ2 debe incluir otras dos observaciones importantes: en primer lugar, un 20% de los portadores de mutaciones no presentan manifestaciones fenotípicas (penetración incompleta); en segundo lugar, la parada cardiaca súbita puede ser la forma de presentación clínica inicial de la enfermedad en hasta un 62% de los casos58. En consecuencia, la TVPC puede considerarse una causa de fibrilación ventricular idiopática mediada por mecanismos adrenérgicos, lo cual justifica la realización de pruebas genéticas en esos casos.
Existen descripciones testimoniales que indican que las mutaciones del KCNJ2 (LQT7) pueden causar un fenotipo de TVPC59. Este punto es de especial importancia en relación con los pacientes negativos para RyR2 y CASQ2, ya que las mutaciones del KCNJ2 suelen asociarse a un pronóstico benigno y la muerte súbita se considera un fenómeno excepcional en estos casos10. Así pues, el diagnóstico molecular puede aportar una perspectiva pronóstica importante en estos casos.
Es muy importante la evaluación genética temprana de todos los familiares de los casos detectados de TVPC, para el diagnóstico presintomático y el consejo adecuado respecto a la reproducción. Además, dado que los bloqueadores beta a menudo son un tratamiento eficaz, el diagnóstico genético de la TVPC tiene interés para la prevención de los episodios con peligro para la vida, puesto que mejora sustancialmente el pronóstico51, y está justificado que pase a ocupar un primer plano en el tratamiento.
Miocardiopatía ventricular derecha arritmogénicaLa MVDA es un trastorno del desmosoma cardiaco, una proteína encargada de mantener la estabilidad estructural a través de la adhesión intercelular, regular la transcripción de los genes que intervienen en la adipogénesis y la apoptosis y mantener la conductividad eléctrica adecuada a través de la regulación de las uniones de hendidura y la homeostasis del calcio60. La prevalencia estimada de la enfermedad es de 1:5.000 y se cree que contribuye de manera importante a producir los casos de MSC de individuos jóvenes y deportistas en todo el mundo, con una tasa anual de mortalidad de un 2-4%61.
La MVDA, una enfermedad predominantemente autosómica dominante, se caracteriza por la degeneración miocárdica y la infiltración fibroadiposa de la pared libre del ventrículo derecho, la región subtricuspídea y el infundíbulo de salida. Se ha descrito también una variante autosómica recesiva muy poco frecuente (enfermedad de Naxos) caracterizada por una afección miocárdica característica, queratosis palmar y pelo lanoso62. La histopatología de la MVDA se caracteriza por la sustitución progresiva del tejido miocárdico por tejido fibroadiposo, predominantemente en el ventrículo derecho, pero que a menudo afecta también al izquierdo (hasta en un 25% de los casos), y que da lugar a una arritmia maligna de origen ventricular63.
Análisis genético en la miocardiopatía ventricular derecha arritmogénicaLa mutación de los genes que codifican alguno de los cinco componentes principales del desmosoma puede conducir a una MVDA, pero los genes PKP2 (que codifica la placofilina 2), DSG2 (que codifica la desmogleína 2) y DSP (que codifica la desmoplaquina) albergan la mayor parte de las mutaciones identificadas: el 27, el 26 y el 11%, respectivamente64. En un análisis conjunto, se identifica una única mutación heterocigota en el 39,2% de los individuos con MVDA en los que se ha realizado un análisis de la secuencia completa de todos los genes del desmosoma65.
Según los criterios revisados de la MVDA de la Task Force66, la identificación de una mutación patógena es el criterio principal para el diagnóstico y resalta la posible utilidad de las pruebas genéticas clínicas en el diagnóstico de la MVDA.
Posibilitar la realización de pruebas de detección en cascada en los familiares pasa a ser la cuestión principal, puesto que un diagnóstico positivo en un familiar modifica la probabilidad de la enfermedad en un individuo en quien se sospeche, lo que hace que pase de entre 1:2 a 1:1.000 y 1:5.00067. Así pues, puede recomendarse la determinación del genotipo en la MVDA para confirmar algunos casos iniciales seleccionados. Se debe examinar clínicamente a los familiares de primer grado mediante ECG de 12 derivaciones, ecocardiografía y resonancia magnética cardiaca. La determinación del genotipo no se ha desarrollado todavía lo suficiente para que pase a ocupar un lugar de primera línea.
Actualmente no hay una clara estratificación del riesgo que pueda derivarse del examen de la determinación del genotipo de la MVDA; la literatura reciente indica que los pacientes positivos para PKP2 sufren síntomas y en ellos la arritmia aparece a edad más temprana, pero la determinación prospectiva de los episodios de activación del desfibrilador no mostraron una diferencia significativa respecto a los pacientes negativos para PKP268.
Limitaciones actuales y papel de las pruebas genéticasDebido al mayor uso de las pruebas genéticas, el consejo genético ha pasado a ser una parte fundamental del proceso. En algunos casos, los efectos beneficiosos obtenidos con la identificación de una mutación patógena pueden ser sustanciales; en otros, la misma identificación podría tener inconvenientes importantes69. El diagnóstico, la prevención, el riesgo de episodios y la respuesta al tratamiento se ven influidos por el genotipo, aun cuando su papel clínico es esencialmente específico para cada enfermedad.
La determinación del genotipo puede no ser apropiada en todos los casos de enfermedad arritmogénica hereditaria, y la decisión de ofrecer o no el análisis debe tomarse en el contexto de cada familia y con su colaboración. Además, la identificación de un número considerable de genes menores que explican pocos casos incrementa la incertidumbre existente en la interpretación. El reciente documento de consenso publicado conjuntamente por la Heart Rhythm Society y la European Heart Rhythm Association ha delimitado claramente los pros y los contras de las pruebas genéticas para cada uno de los trastornos hereditarios70, 71. El documento plantea también el concepto interesante de «genes clave» para referirse a los genes que hay que incluir en una prueba de detección «ideal» de cada trastorno para maximizar la posibilidad de obtener resultados clínicamente útiles y minimizar el riesgo de identificar variantes de trascendencia desconocida, que pueden comportar problemas importantes de interpretación de los resultados de las pruebas genéticas. En la Tabla 3 se resumen las indicaciones de las pruebas genéticas para los trastornos arritmogénicos hereditarios descritos en este artículo y señalados en el documento de consenso.
Tabla 3. Indicaciones para las pruebas genéticas
| Enfermedad | Diagnóstico confirmado/sintomáticos | Asintomáticos | Familiares |
| SQTL | Recomendado: KCNQ1, KCNH2 y SCN5A | Recomendado QT > 500 (480 Ped); en los demás casos puede estar indicado | Recomendado |
| TVPC | Recomendado | Recomendado | Recomendado |
| SBr | Puede ser útil | No indicado si es de tipo 2 o 3 | Recomendado |
| SQTC | Puede considerarse | — | Recomendado |
| MVDA | Puede ser útil | Puede considerarse (1 criterio mayor o 2 criterios menores) | Recomendado |
MVDA: miocardiopatía ventricular derecha arritmogénica; SBr: síndrome de Brugada; SQTC: síndrome de QT corto; SQTL: síndrome de QT largo; TVPC: taquicardia ventricular polimórfica catecolaminérgica.
Durante los últimos 20 años, hemos asistido a un avance increíble en la apreciación del papel de la genética en las arritmias cardiacas. Actualmente disponemos de información cada vez más amplia que plantea nuevos retos y exige mayor interacción entre cardiólogos y especialistas en ciencias básicas. Una vez descubierta una mutación genética, es importante integrar los estudios de laboratorio y los clínicos para determinar su posible efecto nocivo y definir la mejor estrategia para los portadores de la mutación.
La capacidad del equipo de asistencia de determinar la probabilidad de la enfermedad previa a la prueba determina el uso apropiado de la tecnología. En general, estas observaciones resaltan el concepto de que la investigación futura orientada al uso de la información genética para el manejo clínico y terapéutico de los pacientes con una enfermedad arritmogénica hereditaria podría incluir la necesidad de una caracterización funcional, para poder proporcionar una asistencia sanitaria individualizada y específica para el paciente. Nosotros respaldamos la opinión de que la utilidad práctica del análisis genético es diferente en cada trastorno arritmogénico hereditario; está directamente relacionado con el enfoque multidisciplinario en que el cardiólogo, el genetista clínico y el biólogo molecular son coprotagonistas de la actuación médica para alcanzar el mejor uso posible de la información genética. Aunque las pruebas genéticas para las arritmias cardiacas hereditarias no deben ser las primeras que se realicen, está claro que deben ocupar un lugar de primera línea en la práctica clínica actual.
En conjunto, el campo de la genética cardiovascular, de reciente desarrollo, se enfrenta a los futuros retos que comporta llenar las lagunas que separan la práctica clínica actual del tratamiento individualizado de los pacientes.
Conflicto de interesesNinguno.
Autor para correspondencia: Medicina Molecolare, IRCCS Fondazione Salvatore Maugeri, Via Maugeri 10/10A, 27100 Pavía, Italia. silvia.priori@fsm.it