Comité de la ESC para la elaboración de Guías de Práctica Clínica (GPC): Silvia G. Priori (Presidente) (Italia), María Ángeles Alonso García (España), Jean-Jacques Blanc (Francia), Andrzej Budaj (Polonia), Martin Cowie (Reino Unido), Veronica Dean (Francia), Jaap Deckers (Países Bajos), Enrique Fernández Burgos (España), John Lekakis (Grecia), Bertil Lindahl (Suecia), Gianfranco Mazzotta (Italia), Keith McGregor (Francia), João Morais (Portugal), Ali Oto (Turquía) y Otto A. Smiseth (Noruega)
Revisores del documento: Gianfranco Mazzotta (Coordinador de revisión de las CPG) (Italia), Joan Albert Barbera (España), Simon Gibbs (Reino Unido), Marius Hoeper (Alemania), Marc Humbert (Francia), Robert Naeije (Bélgica), Joanna Pepke-Zaba (Reino Unido)
Con permiso de The European Society of Cardiology (ESC).
ÍNDICE DE CONTENIDOS
Preámbulo 524
Introducción 525
Clasificación clínica de la hipertensión pulmonar 526
Hipertensión arterial pulmonar idiopática 527
Factores de riesgo y enfermedades asociadas 527
Enfermedad venooclusiva pulmonar y hemangiomatosis capilar pulmonar 528
Clasificación de los cortocircuitos sistémico-pulmonares congénitos 528
Patología de la hipertensión arterial pulmonar 529
Arteriopatía pulmonar 529
Venopatía oclusiva pulmonar 529
Microvasculopatía pulmonar 529
Patogenia de la hipertensión arterial pulmonar 530
Estrategia diagnóstica 531
Sospecha clínica de hipertensión pulmonar 532
Detección de la hipertensión pulmonar 532
Electrocardiograma 532
Radiografía torácica 532
Ecocardiografía transtorácica con Doppler 532
Identificación de la clase clínica de la hipertensión pulmonar 533
Pruebas de función pulmonar y gasometría arterial 533
Gammagrafía de ventilación-perfusión (V/Q) pulmonar 533
Tomografía computarizada de alta resolución pulmonar 534
Tomografía computarizada espiral pulmonar mejorada con contraste, angiografía pulmonar y resonancia magnética 534
Evaluación de la hipertensión arterial pulmonar (tipo, capacidad de ejercicio, hemodinámica) 534
Analítica sanguínea e inmunología 534
Ecografía abdominal 534
Tolerancia al ejercicio 535
Hemodinámica 535
Biopsia pulmonar 536
Valoración de la severidad 536
Variables clínicas 537
Tolerancia al ejercicio 537
Parámetros ecocardiográficos 538
Hemodinámica 538
Analítica sanguínea 538
Tratamiento 538
Introducción al nivel de evidencia y al grado de recomendación 539
Medidas generales 539
Actividad física 539
Viajes/altitud 539
Prevención de infecciones 539
Embarazo, control de natalidad y terapia hormonal sustitutiva posmenopáusica 541
Concentraciones de hemoglobina 541
Medicación concomitante 541
Asistencia psicológica 541
Cirugía electiva 541
Tratamiento farmacológico 542
Tratamiento anticoagulante oral 542
Diuréticos 542
Oxígeno 542
Digital y dobutamina 543
Bloqueadores de los canales del calcio 543
Prostaciclina sintética y análogos de la prostaciclina 544
Epoprostenol 544
Treprostinil 546
Beraprost sódico 547
Iloprost inhalado 547
Iloprost intravenoso 547
Antagonistas de los receptores de la endotelina 1 547
Bosentán 548
Sitaxsentan 549
Ambrisentan 549
Inhibidores de la fosfodiesterasa tipo 5 550
Sildenafilo 550
Terapia combinada 550
Procedimientos intervencionistas 551
Septostomía auricular con balón 551
Trasplante de pulmón 551
Algoritmo de tratamiento 551
Enfermedades específicas 553
Hipertensión arterial pulmonar pediátrica 553
Hipertensión arterial pulmonar asociada con el síndrome de Eisenmenger 554
Hipertensión portopulmonar 555
Hipertensión arterial pulmonar asociada con la infección por VIH 557
Hipertensión arterial pulmonar asociada con enfermedades del tejido conectivo 558
Enfermedad venooclusiva pulmonar y hemangiomatosis capilar pulmonar 559
Agradecimientos 560
Apéndice A. Lista de abreviaturas 560
Bibliografía 561
PREÁMBULO
Las Guías de Práctica Clínica y los Documentos de Consenso de Expertos tienen como objetivo presentar todas las evidencias relevantes sobre un tema particular para ayudar a los médicos a sopesar los riesgos y los beneficios de un diagnóstico particular o de un procedimiento terapéutico. Deberían ser útiles para la toma diaria de decisiones clínicas.
En los últimos años, la Sociedad Europea de Cardiología (ESC) y otras organizaciones y sociedades relacionadas han elaborado un gran número de Guías de Práctica Clínica y Documentos de Consenso de Expertos. Esta gran profusión puede poner en riesgo la autoridad y validez de las Guías, que sólo pueden estar garantizadas si se han desarrollado mediante un proceso incuestionable de toma de decisiones. Ésta es una de las razones por las que la ESC y otras sociedades han hecho pública una serie de recomendaciones para abordar y formular las Guías de Práctica Clínica y los Documentos de Consenso de Expertos.
A pesar de que los estándares para elaborar las Guías de Práctica Clínica y los Documentos de Consenso de Expertos de calidad están bien definidos, algunas evaluaciones recientes de las Guías de Práctica Clínica y los Documentos de Consenso de Expertos publicadas en revistas con evaluación por pares entre 1995 y 1998 han mostrado faltas en el cumplimiento de los estándares metodológicos en la mayoría de los casos. Por lo tanto, es de la máxima importancia que las Guías y recomendaciones se presenten en formatos que puedan ser fácilmente interpretados. En consecuencia, sus programas de implementación también deben ser correctamente realizados. En este sentido, se han realizado algunos intentos para determinar si las Guías mejoran la calidad de la práctica clínica y la utilización de los recursos sanitarios.
El Comité para las Guías de Práctica Clínica (GPC) de la ESC supervisa y coordina la preparación de nuevas Guías de Práctica Clínica y Documentos de Consenso de Expertos elaborados por los Grupos de Trabajo, grupos de expertos o paneles de consenso. Se solicita a los expertos seleccionados para estos paneles que faciliten una declaración sobre todas sus posibles relaciones que puedan ser consideradas como causa de un conflicto de interés real o potencial. Estos formularios se guardan en forma de ficheros en la Casa Europea del Corazón, la oficina central de la ESC. El Comité es responsable también de la aprobación de estas Guías de Práctica Clínica y Documentos de Consenso de Expertos o de sus comunicados.
El Grupo de Trabajo ha clasificado la utilidad o eficacia del procedimiento y/o tratamiento recomendados y el Nivel de Evidencia tal como se indica en las siguientes tablas:

INTRODUCCIÓN
La hipertensión arterial pulmonar (HAP) se define como un grupo de enfermedades caracterizadas por el aumento progresivo de la resistencia vascular pulmonar (RVP) que conduce al fallo del ventrículo derecho y a la muerte prematura1. Durante la década de los ochenta, cuando aún no se disponía de una terapia específica para esta enfermedad, la esperanza media de vida desde el momento del diagnóstico de los pacientes con HAP idiopática (HAPI), conocida entonces como hipertensión pulmonar primaria (HPP), era de 2,8 años2. La HAP3 incluye la HAPI y la hipertensión pulmonar asociada a distintas afecciones, como enfermedades del tejido conectivo (ETC), cortocircuitos sistémico-pulmonares congénitos, hipertensión portal e infección por el virus de la inmunodeficiencia humana (VIH)4. En todas estas enfermedades están presentes cambios patológicos equivalentes que obstruyen la microcirculación pulmonar5,6 y sugieren que el espectro de enfermedades de la HAP7 comparte procesos biopatológicos.
En la última década hemos asistido a importantes avances en la comprensión del mecanismo de desarrollo de la HAP, su proceso de diagnóstico y su tratamiento.
La identificación de las mutaciones en el receptor 2 de la proteína morfogenética ósea (BMPR2) en la mayoría de los casos de HAP familiar ha constituido un gran avance en la elucidación de la secuencia patogénica de la HAP8,9. Se ha descrito una serie de anomalías celulares en los vasos pulmonares de los pacientes afectados que pueden tener un papel importante en el desarrollo y la progresión de la HAP7. Estas anomalías incluyen la disfunción endotelial pulmonar10, que se caracteriza por una alteración en la síntesis de óxido nítrico (NO), tromboxano A2 (TxA2), prostaciclina y endotelina, alteraciones en los canales de potasio y en la expresión del transportador de la serotonina en las células musculares lisas y un aumento de la producción de matriz extracelular en la adventicia7.
El diagnóstico se define hoy día con más claridad de acuerdo con una nueva clasificación clínica, junto con el consenso alcanzado sobre la aplicación de algoritmos con distintas pruebas diagnósticas y procedimientos que permiten excluir otras etiologías y garantizan un diagnóstico más preciso de la HAP11. Además, se ha propuesto la utilización de marcadores no invasivos de la severidad de la enfermedad, como marcadores bioquímicos y pruebas fisiológicas de amplia disponibilidad para un control fiable de la evolución clínica11,12.
Por último, los numerosos estudios clínicos controlados realizados sobre la HAP nos permiten abandonar la estrategia de tratamiento basada en los datos clínicos y adoptar una terapia basada en la evidencia que incluye nuevos tipos de fármacos, como los prostanoides13, los antagonistas del receptor de la endotelina14 y los inhibidores de la fosfodiesterasa tipo 515.
La presente guía pretende proporcionar indicaciones claras y concisas para la aplicación práctica de la nueva clasificación clínica, además de una breve descripción de la nueva clasificación patológica y de los últimos descubrimientos sobre la patogenia de esta enfermedad. Discutiremos el proceso diagnóstico a efectos de proponer una secuencia lógica de estudios encaminados a identificar la etiología, así como a valorar la enfermedad y su seguimiento. Se pondrá un énfasis especial en el algoritmo de tratamiento basado en la evidencia, que ha sido definido de acuerdo con las propuestas de la ESC sobre la clasificación de los niveles de evidencia y las clases de recomendaciones16 de las terapias disponibles.
CLASIFICACIÓN CLÍNICA DE LA HIPERTENSIÓN PULMONAR
La hipertensión pulmonar (HP) se define como la presencia de una presión media en la arteria pulmonar (PAPm) > 25 mmHg en reposo o > 30 mmHg durante el ejercicio17. En la tabla 1 se presenta la clasificación actual de la HP. Esta clasificación es el resultado de una amplia discusión y representa un consenso que incorpora nuestra comprensión actual de su fisiopatología, así como las diferencias y similitudes de la HP basadas en la evidencia clínica. Las explicaciones que se exponen a continuación pretenden favorecer la comprensión y la correcta aplicación clínica de la nueva clasificación.

La HP se clasificaba anteriormente en 2 categorías: HPP o HP secundaria, dependiendo de la ausencia o presencia de causas identificables o factores de riesgo3,17. El diagnóstico de HPP era de exclusión tras descartar todas las causas de HP.
En 1998, durante el Segundo Congreso Mundial sobre HP celebrado en Evian (Francia), se propuso una clasificación de la HP basada en datos clínicos18. El propósito de la «clasificación de Evian» era individualizar diferentes categorías que compartían similitudes en los mecanismos fisiopatológicos, la presentación clínica y las opciones terapéuticas. Una clasificación de este tipo es esencial a la hora de informar sobre pacientes concretos, para estandarizar el diagnóstico y el tratamiento, para desarrollar estudios con grupos homogéneos de pacientes y para analizar nuevas anomalías biopatológicas en poblaciones de pacientes bien caracterizadas. Obviamente, una clasificación clínica no excluye otro tipo de clasificaciones, como la clasificación patológica basada en hallazgos histológicos o la clasificación funcional basada en la severidad de los síntomas. En 2003, el Tercer Congreso Mundial sobre la HAP celebrado en Venecia (Italia) brindó la oportunidad de valorar el impacto y la utilidad de la clasificación de Evian y de proponer algunas modificaciones.
Se decidió mantener la estructura general y la filosofía de la clasificación de Evian. Sin embargo, se propusieron algunos cambios, en concreto: abandonar el término «hipertensión pulmonar primaria (HPP)» y sustituirlo por «hipertensión arterial pulmonar idiopática (HAPI)», reclasificar la enfermedad venooclusiva pulmonar (EVOP) y la hemangiomatosis capilar pulmonar (HCP), actualizar los factores de riesgo y enfermedades asociadas con la HAP y proponer algunas pautas para mejorar la clasificación de los cortocircuitos sistémico-pulmonares congénitos (tabla 1). El objetivo de estas modificaciones fue hacer que la «clasificación clínica de Venecia» resultara más comprensible y fácil de seguir, y que se utilizara como herramienta habitual de trabajo (*).
(*) En esta guía no se ha incluido una referencia específica al manejo de la hipertensión pulmonar trombembólica crónica (grupo 4). Dada la frecuencia con que se presenta este tipo de hipertensión pulmonar en la práctica clínica, resumimos brevemente las recomendaciones actuales del American College of Chest Physicians1: los pacientes con sospecha de HP tromboembólica crónica deben ser referidos a un centro con experiencia para valorar la viabilidad del tratamiento quirúrgico mediante tromboendarterectomía pulmonar. En los pacientes con obstrucción proximal de los vasos pulmonares (arteria pulmonar principal, lobares y segmentarias), la cirugía es el procedimiento terapéutico de elección. Estas recomendaciones están basadas en la opinión de expertos, ya que no se han realizado ensayos clínicos.
1. Doyle R, Mc Crory D, Channick R, Simonneau G, Conte J. Surgical treatments/interventions for pulmonary arterial hypertension. ACCP Evidence-Based Clinical Guideliness. Chest. 2004;126:S63-71.
Hipertensión arterial pulmonar idiopática
El término HPP se mantuvo en la clasificación de Evian debido al frecuente uso y a la familiaridad de este término, y también porque resultaba emblemático de 50 años de intensa investigación científica y clínica. Sin embargo, el uso del término «primario» facilitaba la introducción del término «secundario», que fue abandonado en la clasificación de Evian debido a que se utilizaba para describir una serie muy heterogénea de enfermedades. A efectos de evitar cualquier confusión, en Venecia se decidió que la primera categoría denominada hipertensión arterial pulmonar (HAP) debería incluir 3 subgrupos principales:
1.1. La hipertensión arterial pulmonar idiopática (HAPI).
1.2. La hipertensión arterial pulmonar familiar (HAPF).
1.3. La hipertensión arterial pulmonar en relación con factores de riesgo o enfermedades asociadas (HAPEA).
Factores de riesgo y enfermedades asociadas
Un factor de riesgo para la HP es cualquier factor o enfermedad que se sospecha que desempeña un papel predisponente o facilitador del desarrollo de la enfermedad. Los factores de riesgo pueden incluir fármacos y sustancias químicas, enfermedades o el fenotipo (edad, sexo). El término «enfermedades asociadas» se utiliza cuando hay un aumento estadísticamente significativo en la incidencia de la HAP en presencia de un factor predisponente dado, sin que se cumpla el «postulado de Koch» para la relación causal. Debido a que el riesgo absoluto de los factores de riesgo conocido para la HAP es en general bajo, la susceptibilidad individual o la predisposición genética posiblemente desempeñen un papel importante. Durante la reunión de Evian en 1998 se categorizaron distintos factores de riesgo y enfermedades asociadas de acuerdo con la importancia de su asociación con la HP y su posible papel causal. Con el término «definitivo» se indica una asociación basada en varias observaciones concordantes que incluyen algún estudio controlado importante o un estudio epidemiológico inequívoco. Con el término «muy probable» se indican varias observaciones concordantes (que incluyen amplias series de casos y estudios) que no son atribuibles a causas identificadas. El término «posible» indica una asociación que se basa en series de casos, registros u opiniones de expertos. Por último, el término «improbale» indica los factores de riesgo de los que se sospechó pero cuya asociación no se pudo demostrar en estudios controlados.
En la tabla 2 se resumen, de acuerdo con el nivel de evidencia, los factores de riesgo y las enfermedades asociadas ya conocidas19, así como los nuevos «posibles» factores de riesgo para la HAP que fueron recientemente identificados en series de casos o en comunicaciones aisladas de casos. Estos nuevos factores de riesgo posibles incluyen enfermedades hematológicas, tales como la asplenia secundaria a la esplenectomía quirúrgica20, la anemia falciforme21, la talasemia-β22 y las enfermedades mieloproliferativas crónicas23 (policitemia vera, trombocitopenia esencial y mielofibrosis con metaplasia mieloide asociada con la leucemia crónica mieloide o el síndrome mielodisplásico). Los factores de riesgo posibles incluyen también enfermedades genéticas o metabólicas raras, tales como la enfermedad de depósito de glucógeno tipo 1a (enfermedad de Von Gierke)24, la enfermedad de Gaucher25 y la telangiectasia hemorrágica hereditaria (enfermedad de Osler-Weber-Rendu)26.

Enfermedad venooclusiva pulmonar y hemangiomatosis capilar pulmonar
En la clasificación de Evian, la EVOP fue incluida en la categoría de hipertensión pulmonar venosa, que se asocia predominantemente con enfermedades del corazón izquierdo, mientras que la HCP se incluyó en el grupo heterogéneo (último grupo) de HP originada por enfermedades que afectan directamente a los vasos pulmonares. Las similitudes en los aspectos histopatológicos y en la presentación clínica, junto con el posible desarrollo de edema pulmonar durante la terapia con epoprostenol, sugieren que estas enfermedades pueden solaparse. De acuerdo con esto, parece lógico incluir la EVOP y la HCP dentro del mismo grupo, más apropiadamente dentro de la HAP. De hecho, la presentación clínica de la EVOP y de la HCP es en general similar a la presentación clínica de la HAPI, y los factores de riesgo o enfermedades asociadas con la HAP y con la EVOP/HCP son también similares e incluyen el espectro de la esclerodermia, la infección por VIH y el uso de agentes anorexígenos. Por lo tanto, en la nueva clasificación clínica (tabla 1), el grupo 1 de la clasificación clínica de la HAP incluye otro subgrupo denominado HAP asociada con compromiso capilar o venoso significativo (clase clínica 1.4).
Clasificación de los cortocircuitos sistémico-pulmonares congénitos
La clasificación propuesta para los cortocircuitos sistémico-pulmonares congénitos toma en consideración tanto el tipo como las dimensiones del defecto, la presencia de anomalías extracardíacas asociadas y el grado de corrección (tabla 3). Todos estos factores son relevantes en el desarrollo de la HP, en la fisiología del síndrome de Eisengmenger y en el pronóstico.

El síndrome de Eisenmenger puede tener su origen en defectos cardíacos congénitos simples o complejos (alrededor del 30% de los casos)27.
Entre los defectos simples, la comunicación interventricular parece ser el más frecuente, seguida por la comunicación interauricular y el ductus arteriosus patente27. Se calcula que el 10% de los pacientes > 2 años con comunicación interventricular de cualquier tipo pueden llegar a desarrollar el síndrome de Eisenmenger, en comparación con el 4-6% de los pacientes con comunicaciones interauriculares28,29. En los pacientes que presentan comunicaciones mayores, la mayor parte de ellos con truncus arteriosus, el 50% de los casos con comunicación interventricular y el 10% de los que tienen comunicación interauricular desarrollarán HAP y enfermedad vascular pulmonar30. Entre los pacientes con comunicación interauricular, los que presentan comunicación de tipo senovenoso tienen una incidencia más elevada de HAP (16%) que los pacientes con una comunicación de tipo ostium secundum (4%)31.
El desarrollo de la HAP con enfermedad vascular pulmonar parece estar relacionado con el tamaño de la comunicación. De hecho, sólo el 3% de los pacientes con comunicación interventricular pequeña o moderada desarrolla HP32,33. Por el contrario, el 50% de los pacientes con comunicaciones grandes (> 1,5 cm de diámetro) estará afectado. En el caso de las comunicaciones pequeñas (comunicación interventricular < 1 cm y comunicación interauricular < 2 cm de diámetro efectivo valorado por ecocardiografía), el papel fisiopatológico exacto del defecto cardíaco en el desarrollo de la HAP se desconoce.
En algunos pacientes se puede detectar HAP severa después de que el defecto cardíaco haya sido corregido con éxito. En muchos de estos casos no queda claro si las lesiones vasculares pulmonares irreversibles estaban presentes antes de la intervención quirúrgica o si la enfermedad vascular pulmonar progresó a pesar de la corrección del defecto. Normalmente, una corrección temprana previene el desarrollo subsiguiente de la HAP.
PATOLOGÍA DE LA HIPERTENSIÓN ARTERIAL PULMONAR
La HAP incluye varias formas de HP de diferentes etiologías pero con una presentación clínica similar y, en muchos casos, con una respuesta parecida al tratamiento médico. Los cambios histopatológicos en varias formas de la HAP son cualitativamente similares5, pero con diferencias cuantitativas en cuanto a la distribución y prevalencia de los cambios patológicos en los distintos componentes del lecho vascular pulmonar (arteriolas, capilares o venas). En el Tercer Simposio Internacional sobre la HAP celebrado en Venecia se propuso la siguiente clasificación patológica actualizada (tabla 4)6.

Arteriopatía pulmonar
Las características histopatológicas de la arteriopatía pulmonar incluyen la hipertrofia de la media, el engrosamiento de la íntima y de la adventicia, y las lesiones complejas.
La hipertrofia de la media es un aumento del área seccional de la media de las arterias pulmonares preacinares e intraacinares. Se debe tanto a la hipertrofia e hiperplasiaas fibras musculares cardíacas como al aumento de la matriz del tejido conectivo y de las fibras elásticas en la media de las arterias musculares.
El engrosamiento de la íntima puede ser laminar concéntrico, excéntrico o concéntrico no laminar. En los ámbitos ultraestructural e inmunohistoquímico, las células de la íntima presentan formaciones de fibroblastos, miofibroblastos y células musculares lisas.
El engrosamiento de la adventicia ocurre en la mayoría de los casos de HAP, pero es más difícil de evaluar.
En cuanto a las lesiones complejas, la lesión plexiforme es la proliferación focal de canales endoteliales alineados por miofibroblastos, células de músculo liso y matriz de tejido conectivo. Estas lesiones se encuentran en lugares de bifurcación arterial o en el origen de una arteria supranumeraria, distales al marcado engrosamiento intimal obliterativo de la arteria de la que tienen su origen. No se ha determinado la frecuencia de las lesiones plexiformes en la HAP. La arteritis puede estar asociada con las lesiones plexiformes y se caracteriza por la necrosis de la pared arterial con insudación fibrinoide e infiltración de células inflamatorias.
Todos estos cambios son típicos de los grupos 1.1 (HAPI), 1.2 (HAPF) y 1.3 (HAPA) de la clasificación clínica (tabla 1).
Venopatía oclusiva pulmonar (también denominada enfermedad venooclusiva pulmonar)
La venopatía oclusiva pulmonar representa un porcentaje relativamente pequeño de los casos de HP; las principales características histopatológicas consisten en la oclusión extensa y difusa de vénulas y venas pulmonares de distinto tamaño. La oclusión de la luz del vaso puede ser sólida o excéntrica. Además, la media puede aparecer engrosada. En la venopatía oclusiva pulmonar se encuentran grandes cantidades de hemosiderina, tanto en el citoplasma de macrófagos alveolares y neumocitos tipo II como en depósitos en el intersticio. Los vasos capilares aparecen gruesos y prominentes, y son tan tortuosos que imitan a la hemangiomatosis capilar pulmonar. Se puede observar el remodelado de las arteriolas pulmonares, con hipertrofia de la media y fibrosis de la íntima. Las lesiones plexiformes y la arteritis fibrinoide no están descritas dentro de la venopatía oclusiva pulmonar. El instersticio pulmonar muestra con frecuencia edema en los septos lobulares, que puede evolucionar a fibrosis intersticial. Los ganglios linfáticos pulmonares y la pleura también están dilatados. Estos cambios son típicos del grupo 1.4.1 (EVOP) de la clasificación clínica (tabla 1).
Microvasculopatía pulmonar (también denominada hemangiomatosis capilar pulmonar)
La microvasculopatía pulmonar es otra enfermedad rara caracterizada por la proliferación capilar localizada en el pulmón. La distribución de la microvasculopatía pulmonar es normalmente panlobar y parcheada. La proliferación capilar anormal infiltra las paredes de las arterias y de las venas, invadiendo las paredes musculares y obstruyendo la luz. En las zonas de proliferación capilar también está presente la hemosid erosis pulmonar, caracterizada por los macrófagos cargados de hemosiderina y neumocitos tipo II. Al igual que en la venopatía oclusiva pulmonar, en la microvasculopatía pulmonar las arterias pulmonares presentan una marcada hipertrofia muscular y engrosamiento de la íntima. Estos cambios son típicos del grupo 1.4.2 (HCP) de la clasificación clínica (tabla 1).
Por último, también hay entidades inclasificables con características histopatológicas atípicas o con muestreo inadecuado de vasos sanguíneos.
PATOGENIA DE LA HIPERTENSIÓN ARTERIAL PULMONAR
Los procesos que desencadenan los cambios patológicos observados en la HAP se conocen de forma imprecisa, aunque hoy día entendemos mejor sus mecanismos. Sabemos que la HAP tiene una biopatología multifactorial que incluye varios procesos bioquímicos y distintos tipos de células. El aumento de la resitencia vascular pulmonar (RVP) está relacionada con distintos mecanismos, tales como la vasoconstricción, el remodelado obstructivo de la pared de los vasos pulmonares, la inflamación y la trombosis.
Se cree que la vasoconstricción es uno de los primeros componentes del proceso de la hipertensión pulmonar34. Se ha relacionado la vasoconstricción excesiva con la función o expresión anormal de los canales de potasio en las células de músculo liso35 y con la disfunción endotelial10. En pacientes con HAP se han observado concentraciones plasmáticas reducidas de una sustancia vasodilatadora y antiproliferativa como el péptido intestinal vasoactivo36.
La disfunción endotelial conduce a la reducción crónica de la producción de vasodilatadores, como el óxido nítrico (NO) y la prostaciclina, al tiempo que aumenta la expresión de vasoconstrictores, como el tromboxano A2 (TxA2) y la endotelina 1 (ET-1)10. Muchos de estos procesos anormales elevan el tono vascular y promueven el remodelado vascular.
El proceso de remodelado vascular pulmonar afecta a todas las capas de la pared del vaso y se caracteriza por cambios proliferativos y obstructivos en distintos tipos de células, incluidas las endoteliales, las musculares lisas y los fibroblastos6,7. Además, en la adventicia aumenta la producción de matriz extracelular, incluidos el colágeno, la elastina, la fibronectina y la tenascina37. La angiopoyetina 1, un factor angiogénico esencial para el desarrollo vascular pulmonar, parece hiperactivado en los casos de HP y se correlaciona directamente con la severidad de la enfermedad38.
Las células inflamatorias y las plaquetas también parecen tener un papel significativo en la HAP. Las células inflamatorias son ubicuas en todos los cambios patológicos de la HAP y las citocinas proinflamatorias están elevadas en el plasma de los pacientes con HAP39. En estos pacientes también se han detectado alteraciones en los procesos metabólicos de la serotonina, una sustancia vasoconstrictora pulmonar que se almacena en las plaquetas40.
En los pacientes con HAP se han demostrado anomalías protrombóticas41 y se ha encontrado trombos tanto en la microcirculación como en las arterias pulmonares elásticas6. De hecho, las concentraciones de fibrinopéptido A, que reflejan la actividad de la trombina42, y las de TxA243 están elevadas en los pacientes con HAP.
A pesar de la identificación de las mutaciones del BMPR2 (receptor 2 de la proteína morfogenética ósea) en la mayoría de los casos de HAP familiar8,9, todavía no se han aclarado las relaciones biopatológicas entre esta anormalidad genética y el desarrollo de la enfermedad de hipertensión vascular pulmonar. Por otra parte, su alta frecuencia en los casos «verdaderos» de HAPI esporádica y su penetrancia reducida en los casos de HAP familiar (sólo el 20% de los portadores de la mutación genética del BMPR2 manifiesta la enfermedad) nos hace pensar que son necesarios mecanismos adicionales para el desarrollo de esta condición. Estos mecanismos podrían ser las mutaciones somáticas secundarias dentro de un proceso inestable de la vía BMPR244, polimorfismos de genes relacionados con la HAP (gen transportador de la serotonina [5HTT]40, el gen de la sintasa del óxido nítrico [ec-NOS]45, el gen de la carbamilfosfato sintasa [CPS]46) o cualquier estímulo capaz de interrumpir el control del crecimiento de las células vasculares pulmonares. Además, puede haber otros genes, posiblemente relacionados con la vía BMP/TGF-βv, todavía por identificar. De hecho, en pacientes con HAP y antecedentes familiares o personales de telangiectasia hemorrágica hereditaria, por ejemplo, con enfermedad de Osler-Weber-Rendu, se han identificado mutaciones en los receptores TGF-βv, en la cinasa 1 análoga al receptor de la activina (ALK-1) y en la endoglina26,47.
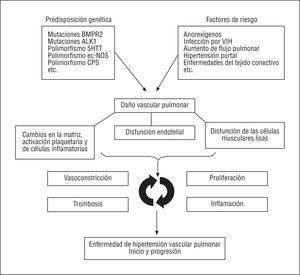
Si bien se han identificado muchos mecanismos biopatológicos en las células y tejidos de los pacientes con HAP, todavía no se comprenden en profundidad las interacciones exactas entre estos mecanismos que dan inicio y hacen progresar los procesos patológicos. Entre las explicaciones teóricas posibles (fig. 1) se incluye la clásica interacción entre predisposición genética y factores de riesgo que puede inducir cambios en diferentes tipos de células (células musculares lisas, endoteliales, inflamatorias y plaquetas) y en la matriz extracelular de la microcirculación pulmonar. El desequilibrio entre factores trombogénicos, mitogénicos, proinflamatorios y vasoconstrictores en contraposición con los mecanismos anticoagulantes, antimitóticos y vasodilatadores puede dar inicio y perpetuar procesos interactivos, como la vasoconstricción, la proliferación, la trombosis y la inflamación de la microcirculación pulmonar. Estos mecanismos son los causantes del inicio y la progresión de los cambios patológicos obstructivos típicos de la HAP. El consiguiente aumento de la RVP conduce a la sobrecarga del ventrículo derecho y, finalmente, a la insuficiencia de éste y a la muerte.

Fig. 1. Hipertensión arterial pulmonar: Mecanismos patogénicos y biopatológicos potenciales. BMPR-2: gen receptor 2 de la proteína morfogenética ósea; ALK 1: gen de la cinasa 1 análoga al receptor de la activina; 5-HTT: gen transportador de la serotonina; ec-NOS: gen de la óxido nítrico sintasa; CPS: gen de la carbamilfosfato sintetasa.
Se necesitan futuros estudios para determinar cuáles de estas alteraciones son las causantes del inicio de la HAP (en caso de que sea alguna) y sobre cuáles debemos actual para curar la enfermedad.
ESTRATEGIA DIAGNÓSTICA
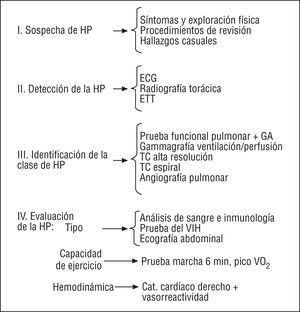
El proceso diagnóstico de la HP requiere una serie de estudios que permitan realizar el diagnóstico, determinar la clasificación clínica de la HP y el tipo de HAP, y evaluar el daño funcional y hemodinámico. Por motivos prácticos puede ser útil seguir una estrategia secuencial que incluye 4 fases (fig. 2):

Fig. 2. Estrategia diagnóstica para la hipertensión pulmonar. GA: gasometría arterial; TC: tomografía computarizada; HP: hipertensión pulmonar; HAP: hipertensión arterial pulmonar; ETT: ecocardiografía transtorácica; VO2: consumo de oxígeno; Cat.: cateterismo.
1. Sospecha clínica de hipertensión pulmonar.
2. Detección de la hipertensión pulmonar.
3. Clasificación clínica de la hipertensión pulmonar.
4. Evaluación de la hipertensión arterial pulmonar (tipo, capacidad funcional, hemodinámica).
Sospecha clínica de hipertensión pulmonar
Debemos sospechar la presencia de HP en cualquier paciente que sienta dificultad al respirar y no se encuentren otros signos claros de enfermedad cardíaca o pulmonar, o en pacientes con enfermedad cardíaca o pulmonar subyacente pero cuando ésta no justifique la disnea progresiva. Entre los síntomas de la HP48 también se pueden incluir el cansancio, la debilidad, la angina, el síncope y la distensión abdominal. Sólo en casos muy avanzados se ha comunicado la presencia de estos síntomas en reposo.
Para apreciar los signos físicos de la HP48 es necesario tener una amplia experiencia clínica. Entre estos signos se incluyen el impulso del borde paraesternal izquierdo, el aumento del componente pulmonar del segundo ruido, un soplo pansistólico de regurgitación tricúspide, un soplo diastólico de insuficiencia pulmonar y un tercer ruido ventricular derecho. Los pacientes en un estadio más avanzado de la enfermedad y con insuficiencia del ventrículo derecho en reposo se caracterizan por la presencia de distensión venosa yugular, hepatomegalia, edema periférico, ascitis y extremidades frías. También se puede observar cianosis central (algunas veces cianosis periférica o ambas). La auscultación pulmonar suele ser normal.
Aparece sospecha clínica cuando se observan estos síntomas y signos en pacientes con enfermedades que pueden estar asociadas con la HAP, tales como enfermedad del tejido conectivo (ETC), hipertensión portal, infección por VIH y enfermedades cardíacas congénitas con cortocircuitos sistémico-pulmonares. En presencia de estos condicionantes predisponentes, algunos especialistas proponen un control médico periódico que permita identificar a los pacientes asintomáticos en la fase temprana de la HP49 (véase el apartado Enfermedades específicas).
Por último, se puede sospechar una HP si en el transcurso de procedimientos realizados por otras razones clínicas se descubren hallazgos anormales mediante electrocardiografía, radiografía torácica o ecocardiografía.
Detección de la hipertensión pulmonar
En la fase de detección de la enfermedad es preciso realizar estudios que confirmen el diagnóstico de HP. Los estudios incluyen el electrocardiograma (ECG), radiografía torácica y ecocardiografía transtorácica con Doppler (ETT).
Electrocardiograma
El ECG puede sugerir o proporcionar evidencia de HP si se observa hipertrofia con sobrecarga del ventrículo derecho y dilatación de la aurícula derecha. La hipertrofia ventricular derecha en el ECG está presente en el 87% y la desviación del eje a la derecha en el 79% de los pacientes con HAPI48. Sin embargo, el ECG tiene una sensibilidad y especificidad inadecuadas (del 55 y el 70%, respectivamente) y no puede ser considerado como la herramienta óptima de control para la detección de la HAP significativa50. Un ECG normal no excluye la presencia de HP severa.
Radiografía torácica
En el 90% de los pacientes con HAPI, la radiografía torácica es anormal en el momento del diagnóstico48. Los hallazgos incluyen dilatación arterial pulmonar central que contrasta con la «amputación» de los vasos sanguíneos periféricos. Se puede observar el aumento auricular y ventricular derechos, que en los casos más avanzados es progresivo. La radiografía torácica permite excluir, razonablemente, la asociación de enfermedad pulmonar moderada o severa o la hipertensión venosa pulmonar debida a alteraciones del corazón izquierdo. Sin embargo, una radiografía torácica normal no excluye la presencia de HP poscapilar leve, que incluye enfermedad del corazón izquierdo o enfermedad pulmonar venooclusiva.
Ecocardiografía transtorácica con Doppler
La ETT es una prueba no invasiva excelente para los pacientes en los que se sospecha HP. Mediante ETT se estima la presión sistólica arterial pulmonar (PSAP) y se puede obtener información adicional sobre la causa y las consecuencias de la HP. La PSAP es equivalente a la presión sistólica del ventrículo derecho (PSVD) en ausencia de obstrucción del tracto de salida pulmonar. La PSVD se puede calcular midiendo la velocidad del flujo regurgitante sistólico tricuspídeo (v) y una estimación de la presión auricular derecha (PAD), aplicadas en la fórmula: PSVD = 4v² + PAD. La presión de la aurícula derecha (PAD) puede tener un valor estándar o un valor estimado según las características de la vena cava inferior51 o a la distensión venosa yugular. El flujo regurgitante tricuspídeo puede ser visualizado en la mayoría (74%) de los pacientes con HP52. En la mayor parte de los estudios se ha observado una elevada correlación (0,57-0,93) entre las mediciones de la PSAP realizadas mediante ETT y con cateterismo cardíaco derecho53. Sin embargo, con el objeto de minimizar falsos positivos54 es importante identificar valores específicos para la definición de HP valorada por ETT.
El rango de PSVD en sujetos sanos ha sido bien caracterizado. En una amplia población de ambos sexos, de 1-89 años de edad, se calculó una PSVD de 28 ± 5 mmHg (rango, 15-57 mmHg). La PSVD se incrementa con la edad y con el índice de masa corporal55. De acuerdo con estos datos, la HP leve se puede definir como una PSAP de aproximadamente 36-50 mmHg o una velocidad de la regurgitación tricúspide en reposo de 2,8-3,4 m/s (asumiendo una PAD normal de 5 mmHg). Cabe destacar que incluso con esta definición se pueden anticipar una serie de diagnósticos falsos positivos, especialmente en personas de edad avanzada, y es precisa la confirmación mediante cateterismo cardíaco derecho en pacientes sintomáticos (insuficiencia cardíaca de clase II-III de la NYHA). En sujetos asintomáticos (clase I de la NYHA) hay que excluir una enfermedad del tejido conectivo (ETC) concomitante y se debe repetir la ecocardiografía a los 6 meses. Es preciso señalar también que al definir el nivel de una PSVD elevada, no se define el punto en el que el incremento de la PSVD es clínicamente importante, es un predictor de futuras consecuencias y/o requiere tratamiento específico. Además, ante la posibilidad de un falso negativo en la ecocardiografía Doppler se debe considerar la posibilidad de HP si la sospecha clínica es importante56 (*).
(*) Un algoritmo en el manejo de los pacientes con hipertensión pulmonar ligera (PASP estimada de 36-50 mmHg) es muy útil, dada la alta frecuencia de este hallazgo en la práctica clínica. Los pacientes asintomáticos con hipertensión pulmonar ligera deben ser evaluados periódicamente (cada 6 meses), con un nuevo ecocardiograma. En los pacientes sintomáticos con hipertensión pulmonar ligera se realizará un cateterismo derecho para confirmar el diagnóstico.
Para la confirmación del diagnóstico y la valoración de la severidad de la HP son importantes los parámetros ecocardiográficos y Doppler, incluidos la dimensión y función del ventrículo derecho e izquierdo, la presencia de anormalidades en las válvulas tricúspide, pulmonar y mitral, la eyección del ventrículo derecho y las características de llenado del ventrículo izquierdo, las dimensiones de la vena cava inferior y el tamaño de un derrame pericárdico57,58.
Aparte de la identificación de la HP, la ETT permite establecer un diagnóstico diferencial al determinar posibles causas, lo que da inicio a las fases III y IV del proceso diagnóstico. La ETT permite identificar enfermedades de las válvulas del corazón izquierdo y enfermedades miocárdicas, causantes de la hipertensión venosa pulmonar (clase clínica 2), y enfermedades cardíacas congénitas con cortocircuito sistémico-pulmonar (clase clínica 1.3.2). La inyección intravenosa de suero salino agitado, utilizado como medio de contraste, puede ayudar a identificar un foramen oval permeable o una comunicación interauricular tipo seno venoso pequeña que pudiesen escapar a su detección mediante ETT convencional. Rara vez se requiere ETT, que normalmente se utiliza para confirmar la presencia y valorar el tamaño exacto de pequeños defectos del septo auricular.
Identificación de la clase clínica de la hipertensión pulmonar
El siguiente paso tras la detección de la HP es la identificación de la clase clínica de acuerdo con la clasificación clínica de Venecia (tabla 1)1. Esto puede realizarse a través del uso de pruebas esenciales, tales como la ETT, tal y como se ha especificado anteriormente, las pruebas de función pulmonar (PFP) (que incluyen la gasometría arterial) y las pruebas de ventilación-perfusión. En caso necesario, podrían realizarse pruebas adicionales en circunstancias particulares, tales como la tomografía computarizada de alta resolución (TCAR) de tórax, la tomografía computarizada (TC) espiral y la angiografía pulmonar.
Pruebas de función pulmonar y gasometría arterial
Las PFP y gasometrías arteriales pueden identificar la contribución de enfermedades subyacentes, tanto de las vías aéreas como parenquimatosas. Los pacientes con HAP tienen normalmente una capacidad de difusión disminuida para el monóxido de carbono (DLCO) en los pulmones (típicamente en el rango del 40-80% del predecible) y una reducción leve o moderada de los volúmenes pulmonares. La presión arterial de oxígeno (PaO2) es normal o sólo ligeramente más baja de lo normal y la presión arterial de dióxido de carbono (PaCO2) se encuentra disminuida como resultado de la hiperventilación alveolar. La enfermedad pulmonar obstructiva crónica causante de la HP hipóxica se diagnostica con la evidencia de la obstrucción irreversible de las vías aéreas59, que normalmente se mide a través del volumen expiratorio forzado por segundo (FEV1). Estos pacientes tienen una PaCO2 normal o aumentada, junto con limitación de las vías aéreas y un incremento de los volúmenes residuales, así como una DLCO reducida. El enfisema se diagnostica actualmente mediante la TCAR. La disminución en el volumen pulmonar, junto con la disminución en el DLCO, puede llevar al diagnóstico de enfermedad pulmonar intersticial (EPI). Una vez más, la TCAR de tórax es el principal método para valorar la severidad de la EPI60. Si se sospecha clínicamente, la monitorización oximétrica durante la noche y la polisomnografía permitirán excluir una apnea/hipoapnea obstructiva del sueño y desaturación nocturna.
Gammagrafía de ventilación-perfusión (V/Q) pulmonar
En la HAP, la gammagrafía pulmonar V/Q puede ser completamente normal. Sin embargo, también puede mostrar pequeños defectos de perfusión, no segmentarios y periféricos. Éstos tienen una ventilación normal y, por lo tanto, representan una disparidad V/Q. La gammagrafía de ventilación-perfusión pulmonar proporciona el método de diagnóstico de la hipertensión pulmonar por tromboembolia crónica (HPTEC, clase clínica 4)61. En la HPTEC, los defectos de perfusión se encuentran normalmente en las regiones lobares y ocasionan defectos segmentarios en la imagen de perfusión. Si estas zonas se encuentran ventiladas de forma normal, los defectos de perfusión se describen como disparidades en relación con los defectos de ventilación. La gammagrafía de ventilación-perfusión mostró una sensibilidad del 90-100% con una especificidad del 94-100% para distinguir entre la HAPI y la HPTEC61. Los defectos de perfusión con ventilación normal también pueden observarse en la enfermedad venooclusiva. Estos pacientes requieren una cuidadosa investigación adicional (véase el apartado TCAR). En pacientes con enfermedad pulmonar parenquimatosa, los defectos de perfusión son parejos a los defectos de ventilación.
Tomografía computarizada de alta resolución pulmonar
La TCAR pulmonar permite obtener imágenes muy detalladas del parénquima pulmonar y facilita el diagnóstico de la EPI y del enfisema. La presencia de marcadores instersticiales similares a los observados en el fallo ventricular izquierdo avanzado, tales como opacificaciones difusas centrales en forma de reloj de arena y engrosamiento del septo interlobular, sugieren la presencia de enfermedad venooclusiva pulmonar; son hallazgos adicionales, en este caso, la linfadenopatía y el engrosamiento y el derrame pleural62. El engrosamiento bilateral difuso del septo interlobular y la presencia de opacidades pequeñas, centrilobulares y mal circunscritas sugieren la presencia de hemangiomatosis capilar pulmonar.
Tomografía computarizada espiral pulmonar mejorada con contraste, angiografía pulmonar y resonancia magnética
La TC espiral (o helicoidal) mejorada con contraste está indicada en los pacientes con HP que presentan una gammagrafía pulmonar de ventilación-perfusión en la que se encontraron defectos segmentarios o subsegmentarios de perfusión con una ventilación normal (p. ej., evidencia de disparidad V/Q), y puede mostrar la presencia de tromboémbolos centrales pulmonares crónicos. En la TC, la enfermedad tromboembólica crónica se manifiesta con la oclusión completa de las arterias pulmonares, la presencia de defectos de llenado excéntricos compatibles con trombos, la recanalización y la presencia de estenosis o redes vasculares63,64.
La angiografía pulmonar convencional todavía es necesaria en el diagnóstico de la HPTEC para una mejor identificación de los pacientes que pueden beneficiarse de una intervención con endarterectomía61. La angiografía pulmonar es más precisa en la identificación de obstrucciones distales y también está indicada en casos en los que la TC espiral mejorada con contraste proporciona resultados no concluyentes en pacientes con una sospecha clínica y basada en la gammagrafía pulmonar de HPTEC. Este procedimiento puede ser realizado de forma segura por personal médico experimentado en los pacientes que presentan HP severa. Algunos aspectos técnicos de utilidad incluyen el uso de medios de contraste modernos, la inyección selectiva en las ramas principales izquierda y derecha, y la realización de múltiples angulaciones.
La resonancia magnética (RM) se usa de forma creciente en pacientes con HAP para valorar los cambios patológicos y funcionales, tanto en el corazón como en la circulación pulmonar63. Sin embargo, se necesita experiencia adicional antes de incluir esta herramienta en la valoración sistemática de los pacientes con HAP.
Evaluación de la hipertensión arterial pulmonar (tipo, capacidad de ejercicio, hemodinámica)
Una vez se ha establecido la clase clínica de la HAP (clase clínica 1), puede ser necesario realizar estudios adicionales para la identificación exacta del tipo de HAP, así como para la valoración de la capacidad funcional y la hemodinámica.
Analítica sanguínea e inmunología
Es preciso realizar un estudio sistemático de la bioquímica sanguínea, la hematología y la función tiroidea. Debería estudiarse también una potencial trombofilia que incluya la determinación de anticuerpos antifosfolípidos (anticoagulante lúpico, anticuerpos anticardiolipina). Las ETC se diagnostican de forma primaria según criterios clínicos y de laboratorio; un panel de pruebas para la autoinmunidad consiste en la determinación de anticuerpos antinucleares (AAN) que incluya la determinación de anticuerpos anticentrómero, anti-SCL70 y RNP. Alrededor de un tercio de los pacientes con HAPI es positivo, pero presenta una positividad con bajo título de anticuerpos antinucleares (diluciones ≤ 1:80)65. Los pacientes con una elevación sustancial del título de AAN y/o datos clínicos de sospecha requieren una valoración serológica adicional y una interconsulta con reumatología. Por último, en todos los pacientes debe obtenerse el permiso para realizar una serología del VIH.
Ecografía abdominal
Mediante la ecografía abdominal puede excluirse de forma fiable la presencia de cirrosis hepática y/o de hipertensión portal. El estudio con Doppler color también permite diferenciar entre la hipertensiónsecundaria a fallo cardíaco derecho, y la hipertensión portal que tiene su origen en el incremento del gradiente venoso transhepático asociado con la cirrosis hepática. El uso de agentes de contraste puede mejorar el diagnóstico66. La hipertensión portal puede confirmarse tras la detección de un gradiente aumentado de la presión entre las venas hepáticas libres y ocluidas (en cuña o enclavamiento) en el momento del cateterismo cardíaco derecho (CCD) (véase el apartado Hipertensión portopulmonar)67.
Tolerancia al ejercicio
La valoración objetiva de la capacidad de ejercicio en pacientes con HAP es un instrumento importante para valorar la severidad de la enfermedad68,69 y el efecto del tratamiento70,71. Las pruebas de ejercicio más utilizadas para la HP son la prueba de la marcha de 6 min y la prueba de esfuerzo cardiopulmonar con medida de intercambio gaseoso.
La prueba de la marcha de 6 min es técnicamente muy simple y poco costosa72. Permite predecir la supervivencia de la HAPI y, además, guarda una correlación inversa con la severidad de la clase funcional de la NYHA68. La prueba de la marcha de 6 min se combina normalmente con la escala de Borg, que valora el grado subjetivo de disnea durante el ejercicio. Una reducción de la saturación arterial de oxígeno > 10% durante la prueba de la marcha de 6 min se asocia con un incremento del riesgo de mortalidad 2,9 veces durante un período medio de 26 meses73. La prueba de la marcha de 6 min es el parámetro de valoración primario tradicional para la gran mayoría de los ensayos clínicos controlados realizados en el campo de la HAP70.
La prueba de esfuerzo cardiopulmonar (PECP) permite medir la ventilación y el intercambio gaseoso durante el ejercicio, y proporciona información «fisiopatológica» adicional a la que se obtiene con la prueba de esfuerzo convencional. Los pacientes con HAP muestran un pico de consumo de O2 (VO2) reducido, una disminución de la tasa de pico de ejercicio, una reducción del aumento del consumo de oxígeno al aumentar el trabajo, un umbral anaeróbico disminuido y una reducción del pico de pulso de oxígeno. Además, también muestran una pendiente aumentada de VE y VCO2 representativas de una ineficiencia ventilatoria69. El pico de VO2 guarda una relación con el pronóstico de los pacientes con HAP69.
La PECP se ha utilizado en los estudios multicéntricos más recientes, pero no ha sido capaz de confirmar las mejorías observadas en la prueba de la marcha de 6 min74,75. Una explicación posible a estos hallazgos es que la PECP resulta técnicamente más difícil que la prueba de la marcha de 6 min y que sus resultados pueden estar influidos por la experiencia de los centros. Una explicación alternativa podría encontrarse en la falta de sensibilidad de la PECP para la medición de la respuesta al tratamiento por tener menos efecto sobre el ejercicio máximo, en relación con el ejercicio submáximo.
Hemodinámica
El cateterismo cardíaco derecho (CCD) debe realizarse para confirmar el diagnóstico de HAP, para valorar la severidad del impacto hemodinámico y para medir la vasorreactividad de la circulación pulmonar. Deberían siempre valorarse los siguientes parámetros: frecuencia cardíaca, PAD, presión arterial pulmonar (PAP) (sistólica, diastólica y media), presión de enclavamiento capilar pulmonar (PCP), gasto cardíaco (por termodilución o por el método de Flick en casos de cortocircuitos sistémico-pulmonares), presión arterial, resistencia vascular pulmonar (RVP) y sistémica (RVS), saturación arterial y venosa mixta (y saturación en vena cava superior en caso de cortocircuitos sistémico-pulmonares).
La HAP se define como una PAP media > 25 mmHg en reposo o > 30 mmHg durante el ejercicio, por una PCP ≤ 15 mmHg y por una RVP > 3 mmHg/l/min (unidades Wood). El cateterismo cardíaco izquierdo es necesario en las raras circunstancias en las que no puede estimarse el PCP de forma segura.
La confirmación del diagnóstico mediante CCD se hace necesaria en los pacientes sintomáticos (clase II y III de la NYHA) con HP leve estimada mediante ecocardiografía Doppler (la definición se ha expuesto con anterioridad) para identificar a los sujetos que precisan otros procedimientos diagnósticos y terapéuticos adicionales. La valoración de la PCP puede ayudar a distinguir entre la HP arterial y venosa en los pacientes que presentan enfermedades concomitantes del corazón izquierdo.
El CCD es importante también en pacientes que presentan evidencia concluyente de HAP de moderada a severa, ya que las variables hemodinámicas pueden tener una relevancia pronóstica2.
Una elevación en la PAD y en la PAP medias con un gasto cardíaco y una saturación venosa central reducidos permite identificar a los pacientes con HAPI que tienen el peor pronóstico. Las mediciones hemodinámicas se han utilizado también para valorar la historia natural de la HAPI en un paciente individual mediante el uso de una ecuación predictiva2, que también ha sido utilizada para valorar los efectos a largo plazo de nuevos tipos de tratamiento sobre la supervivencia76-78. En cualquier caso, esta fórmula está basada en pacientes tratados con terapia convencional hace unos 15-20 años y, por lo tanto, es posible que no constituyan un grupo de «control» adecuado para las poblaciones de HAP actuales.
Los estudios no controlados sugieren que la administración a largo plazo de bloqueadores de los canales del calcio (BCC) prolonga la supervivencia en los pocos casos de pacientes que responden de forma aguda frente a los pacientes que no presentan tal respuesta79. Por lo general, se acepta que los pacientes que se beneficiarán de un tratamiento prolongado con BCC pueden ser identificados mediante un test de vasodilatación aguda realizado durante el CCD80. Sin embargo, se ha propuesto que la identificación definitiva de los pacientes que pueden beneficiarse del tratamiento con BCC requiere tanto la demostración de una respuesta vasorreactiva aguda positiva como la confirmación de una respuesta sostenida a largo plazo al tratamiento con BCC81.
Las pruebas de vasodilatación aguda deberían hacerse únicamente mediante vasodilatadores pulmonares de acción rápida durante el CCD inicial en centros con experiencia para minimizar los riesgos potenciales82. En la actualidad, los fármacos utilizados en las pruebas agudas son la prostaciclina y la adenosina intravenosa, así como el óxido nítrico inhalado83,84. En la tabla 5 se resumen la vida media, las dosis adecuadas, el incremento y la duración de la administración para dichos compuestos.

La respuesta vasorreactiva aguda positiva (pacientes con respuesta aguda positiva) se define como una reducción de la PAP media ≥ 10 mmHg para alcanzar un valor absoluto de PAP media ≤ 40 mmHg con un gasto cardíaco aumentado o sin cambios11,81,85. En general, sólo un 10-15% de los pacientes con HAPI cumplirá estos criterios81,83. Los pacientes con respuesta aguda positiva presentan una mayor posibilidad de mostrar una respuesta sostenida al tratamiento a largo plazo con dosis altas de BCC, y son los únicos que pueden ser tratados de forma segura con este tipo de fármacos. El tratamiento empírico con BCC sin la realización previa del test vasodilatador agudo es totalmente desaconsejable debido a los posibles efectos adversos severos.
Los pacientes con respuesta positiva a largo plazo al tratamiento con dosis altas de BCC se definen como los que se encuentran en la clase funcional I o II de la NYHA, con valores hemodinámicos prácticamente normales tras varios meses en tratamiento sólo con BCC. Cerca de la mitad de los pacientes con HAPI y respuesta aguda positiva mantienen la respuesta positiva a largo plazo81 al tratamiento con BCC y únicamente en estos casos se precisa su continuación como único tratamiento (*).
(*) El paciente con hipertensión pulmonar que se beneficia del tratamiento crónico con antagonistas del calcio requiere: a) una respuesta positiva en el test vasodilatador agudo, con una reducción de la presión pulmonar media mayor de 10 mmHg, alcanzando un valor absoluto < 40 mmHg; b) verificar la eficacia sostenida en la reducción de las presiones pulmonares tras varios meses de tratamiento crónico con altas dosis de antagonistas del calcio. Previamente1, se seleccionaba a los pacientes para el tratamiento con antagonistas del calcio según la respuesta al test agudo vasodilatador, y se consideraban respondedores si la presión pulmonar media disminuía 10 mmHg sin que fuera necesario alcanzar valores absolutos tan bajos. Sin embargo, en el seguimiento de los pacientes «respondedores» en el test hemodinámico se observó un 50% de fracasos terapéuticos; por ello se han incrementado los valores de exigencia hemodinámica para considerar al paciente respondedor y se ha protocolizado la necesidad de revaluar la eficacia del tratamiento con antagonistas del calcio a los 3-6 meses.
1. Saenz de la Calzada C, Sánchez Sánchez V, Velázquez Martin T, Tello de Meneses R, Gómez Sanchez MA, Delgado Jiménez J, et al. Guías de práctica clínica de la Sociedad Española de Cardiología en tromboembolismo e hipertensión pulmonar. Rev Esp Cardiol. 2001;54:194-210.
La utilidad del test vasodilatador agudo y del tratamiento a largo plazo con BCC en pacientes con HAP asociada a enfermedades subyacentes, tales como ETC o enfermedad cardíaca congénita, es menos clara que con la HAPI81,86. De todas formas, los expertos sugieren que también en estos casos se realice el test vasodilatador agudo y se busque una respuesta a largo plazo a los BCC en los sujetos adecuados.
Biopsia pulmonar
La biopsia pulmonar abierta con toracoscopia entraña riesgos sustanciales de morbilidad y mortalidad. Sobre la base de las escasas posibilidades de modificación del diagnóstico o el tratamiento, se desaconseja la realización sistemática de biopsias (*).
(*) La biopsia pulmonar no forma parte de la evaluación diagnóstica habitual de los pacientes con hipertensión pulmonar; sin embargo, se recomienda su realización cuando se requiere confirmación histológica1 para establecer el diagnóstico de vasculitis pulmonar activa, enfermedad intersticial pulmonar, enfermedad venooclusiva o hemangiomatosis capilar pulmonar cuando han fracasado otras técnicas diagnósticas de menor riesgo para el paciente y hay un alto grado de sospecha clínica.
1. McGoon M, Gutterman D, Steen V, Barst R, Mc Crory D, Fortín T, et al. Screening, early detection and diagnosis of pulmonary arterial hypertension. ACCP Evidence-Based Clinical Guideliness. Chest. 2004;126:S14-34.
VALORACIÓN DE LA SEVERIDAD
Se ha demostrado el valor predictivo de distintas variables en el pronóstico de la HAPI cuando éstas se evalúan en situación basal o tras la instauración de tratamientos específicos71. Se dispone de muy poca información acerca de otras enfermedades, como la HAP asociada a ETC, los cortocircuitos sistémico-pulmonares congénitos, la infección por VIH o la hipertensión portal. En estas circunstancias puede haber factores adicionales que contribuyan al resultado global. De hecho, los pacientes con HAP asociada con ETC tienen peor pronóstico que los pacientes con HAPI, mientras que los pacientes con HAP asociada con cortocircuitos sistémico-pulmonares congénitos presentan una evolución más lenta de la enfermedad que los pacientes con HAPI.
En la práctica, el valor pronóstico de una variable aislada en un paciente individual puede ser menor que el valor de múltiples variables concordantes (tabla 6).

Variables clínicas
Entre las variables clínicas, la clase funcional inicial de la NYHA tiene un valor predictivo pronóstico definitivo en los pacientes con HAPI que reciben tratamiento convencional2. Este valor predictivo se mantiene cuando la clasificación de la NYHA se establece antes o 3 meses después de la iniciación del tratamiento con epoprostenol77,87. Los antecedentes de fallo cardíaco derecho previo a la iniciación del tratamiento con epoprostenol tiene un valor predictivo negativo87. La clasificación de la Organización Mundial de la Salud (OMS) propuesta en Evian es una adaptación del sistema de la NYHA para la HAP y muchos especialistas se refieren a ambas clasificaciones, que son prácticamente idénticas, como clasificación funcional NYHA/OMS (tabla 7)11,12.

Tolerancia al ejercicio
Varios autores han demostrado que la prueba de la marcha de 6 min tiene un gran valor pronóstico en la HAP. Miyamoto et al68 demostraron que los pacientes con HAPI que caminan menos de 332 m presentan una tasa más baja de supervivencia que los que caminan distancias mayores. En otro estudio se estimó que hay una reducción del 18% en el riesgo de muerte por cada 50 m que se caminen en los pacientes con HAPI73. Se dispone de datos preliminares que demuestran que una desaturación arterial de oxígeno > 10% durante la prueba de la marcha de 6 min incrementa el riesgo de mortalidad 2,9 veces en un período medio de seguimiento de 26 meses73. Los pacientes en clase funcional III o IV de la NYHA que caminan ≤ 250 m antes del comienzo del tratamiento con epoprostenol o < 380 m después de 3 meses con tratamiento con epoprostenol tienen un peor pronóstico que los que caminan distancias mayores87. No se ha demostrado que el cambio absoluto de distancia cubierta en el test de los 6 min con epoprostenol tenga un valor pronóstico.
Un VO2 pico < 10,4 ml/kg/min valorado mediante la prueba de esfuerzo cardiopulmonar (PECP) se asocia con un pronóstico peor en los pacientes con HAP69.
Parámetros ecocardiográficos
La presencia y la cuantía del derrame pericárdico valorado por ETT tienen una clara relevancia pronóstica en los pacientes con HAPI88,89. Además, el tamaño de la aurícula derecha y el índice de excentricidad ventricular izquierda son predictivos del curso evolutivo de los pacientes con HAPI89.
Se ha demostrado que el índice ventricular derecho Doppler90 como, por ejemplo, el índice Tei, es una variable que permite valorar tanto la función sistólica como diastólica del ventrículo derecho y tiene relevancia pronóstica en la HAP91.
Hemodinámica
La elevación basal de la PAD y de la PAP medias, así como un gasto cardíaco y una saturación venosa central reducidos, permiten identificar a los pacientes con HAPI con el peor pronóstico2. Los pacientes con respuesta aguda positiva al test vasodilatador agudo tienen mejor pronóstico que los que no la tienen79,83,92.
En el análisis univariable, las variables hemodinámicas en reposo que se asocian con una peor evolución de los pacientes con HAPI, que son posteriormente tratados con epoprostenol, son una PAD > 12 mmHg y una PAP media < 65 mmHg87, si bien este último hallazgo no ha sido confirmado en otras series77. Después de 3 meses de tratamiento con epoprostenol, un descenso en la resistencia vascular pulmonar (RVP) que sea < 30% en relación con los valores basales se asocia con un peor pronóstico87.
Analítica sanguínea
La hiperuricemia ocurre con mucha frecuencia en pacientes con HP y guarda relación con anormalidades hemodinámicas, por ejemplo, la elevación de la PAD y una mayor mortalidad en la HAPI93. El péptido natriurético cerebral se encuentra elevado en casos de sobrecarga de presión ventricular derecha y se correlaciona con la severidad de la disfunción ventricular derecha y la mortalidad en la HAPI94.
Otras concentraciones plasmáticas de neurohormonas guardan relación con la supervivencia, por ejemplo, la noradrenalina95 y la ET-196. Recientemente se ha demostrado que las concentraciones de troponina97, tanto en determinaciones basales como tras la instauración de tratamientos específicos, tienen relevancia pronóstica en pacientes con HAP.
TRATAMIENTO
El tratamiento de la HAP se ha caracterizado tradicionalmente por el escaso número de opciones terapéuticas y las dificultades asociadas en éstas98. Recientemente hemos asistido a un cambio drástico de la situación, tras el lento progreso de las décadas anteriores con la realización de un notable número de ensayos clínicos aleatorizados (ECA) en los últimos años. Sin embargo, nos encontramos ahora con una serie de tratamientos diferentes cuya eficacia se acepta de forma general (p. ej., anticoagulantes orales, oxígeno, BCC), aunque dichos tratamientos no se apoyan en los ECA y no están aprobados formalmente por las autoridades reguladoras como tratamientos específicos indicados para la HAP.
El objetivo de esta sección es revisar cada forma de tratamiento de acuerdo con la clasificación del Nivel de Evidencia sugerido por la Guía de Práctica Clínica (GPC) de la ESC16. Además, proporcionaremos la Clase de Recomendación16, que se refiere a la eficacia clínica de los tratamientos que, por diferentes motivos, pueden no haber sido probados en los ECA, como los anticoagulantes orales, el oxígeno, los BCC, la septostomía auricular con balón y/o el trasplante pulmonar. También proporcionamos información sobre la situación actual relativa a la aprobación, normativa e indicación de cada componente en los distintos países. Por último, se propone un algoritmo de tratamiento basado en la evidencia85 con el que pretendemos proporcionar una guía para el uso selectivo de cada tipo de terapia (*).
(*) La regulación actual por las autoridades sanitarias españolas es: 1. epoprostenol está aceptado para la hipertensión arterial pulmonar idiopática en clase III-IV de la NYHA. 2. Bosentán: se ha asumido la regulación establecida por la EMEA (Agencia Europea para la Evaluación de Productos Médicos). 3. Iloprost: aceptado por la EMEA y pendiente de su aprobación en España que se producirá en los próximos meses. 4. Treprostinil pendiente de aprobación por la EMEA y por las autoridades sanitarias españolas. 5. El resto de los fármacos está pendiente de su aprobación por las distintas agencias del medicamento.
INTRODUCCIÓN AL NIVEL DE EVIDENCIA Y AL GRADO DE RECOMENDACIÓN
El sistema de clasificación de los niveles de evidencia está basado fundamentalmente en el número de ECA realizados sobre una estrategia de tratamiento determinada16 (tabla 8) y adaptados a los requisitos específicos de una enfermedad rara. La única diferencia consiste en que no hemos incluido en la categoría B los «estudios no aleatorizados» ya que, en el caso de la HAP, el número de este tipo de estudios es relativamente pequeño y, por lo tanto, se encuentran incluidos en la categoría C. En la categoría B se incluyen los «estudios clínicos aleatorizados múltiples con resultados heterogéneos», ya que esta situación se puede dar (y se ha dado) y porque esta definición es más amplia, aun cuando el resultado final sea «un solo estudio aleatorizado» con resultados positivos. El análisis tiene en consideración los estudios y ECA realizados en pacientes con HAP publicados en las revistas científicas o presentados en los congresos importantes más recientes. El sistema de elaboración de los niveles de evidencia basado en el número de ECA puede presentar algunas limitaciones que deben ser tenidas en cuenta y, posiblemente, corregidas99. De hecho, el nivel de evidencia puede cambiar con el tiempo como resultado de la realización de estudios adicionales, y el sistema de clasificación de los niveles de evidencia no tiene en cuenta el tamaño de la muestra, por lo que los ensayos clínicos «pequeños» tienen el mismo peso que los «grandes». Además, el nivel de evidencia no debe confundirse con el nivel de eficacia clínica, que depende de los efectos fármaco-dinámicos netos de los componentes y de los posibles efectos secundarios y otras limitaciones (p. ej., la complejidad de la vía de administración). Por ejemplo, una estrategia de tratamiento con mejores resultados pero con uno o ningún ECA se clasifica en la categoría B o C, respectivamente, frente a otra terapia con peores resultados y más efectos secundarios, pero evaluada en más de un ECA, que puede estar en la categoría A. Las autoridades reguladoras pueden aprobar un determinado tratamiento sobre la base de un solo ensayo clínico con un tamaño apropiado de muestra y que cumple adecuadamente con los requisitos estadísticos específicos.

En consecuencia, los grados de recomendación (tabla 9) están basados en el nivel de eficacia clínica que se espera de un procedimiento terapéutico.

Por último, ofrecemos tanto el grado de recomendación como el nivel de evidencia para proporcionar un perfil completo de cada tratamiento (tabla 10). No se recoge el grado de recomendación de los fármacos cuya utilización actual está limitada solamente a los pacientes participantes en ensayos clínicos. En la tabla 11 se recoge el estado de aprobación y etiquetado de cada componente por países.


Medidas generales
Las medidas generales incluyen estrategias dedicadas a disminuir el impacto deletéreo de algunas circunstancias y agentes externos en los pacientes con HAP. Al igual que sucede con otras condiciones clínicas, el impacto de estas medidas no ha sido probado científicamente y, por tanto, las recomendaciones están basadas en la opinión de expertos en la materia.
Recomendación de clase IIa; nivel de evidencia C.
Actividad física
No está claro si la actividad física puede tener un impacto negativo en la evolución de la HAP. Sin embargo, algunos síntomas potencialmente peligrosos, como la disnea severa, el síncope y el dolor torácico, deberían ser evitados. El ejercicio debería limitarse a una situación libre de síntomas con el fin de mantener en buenas condiciones la musculatura del esqueleto. Se debe evitar la actividad física después de las comidas o ante temperaturas extremas. Un adecuado ajuste de las actividades diarias puede mejorar la calidad de vida y reducir la frecuencia de los síntomas.
Viajes/altitud
La hipoxia puede agravar la vasoconstricción en los pacientes con HAP y es conveniente evitar grados leves de hipoxia hipobárica que comienzan en altitudes entre 1.500 y 2.000 m. Los vuelos comerciales están presurizados a una altitud equivalente a 1.600-2.500 m y debería considerarse el uso de oxígeno suplementario en los pacientes con HAP. Antes de planificar un viaje, el paciente debe consultar al especialista en HP.
Prevención de infecciones
Los pacientes con HAP tienen tendencia a desarrollar neumonía, que es la causa de muerte en el 7% de los casos. Las infecciones pulmonares se toleran muy mal y es necesario que sean diagnosticadas y tratadas de forma precoz. Se recomienda una estrategia de vacunación para la gripe y para la neumonía neumocócica. La fiebre persistente en pacientes con catéteres intravenosos para la administración continua de epoprostenol debe hacer sospechar la infección del catéter.
Embarazo, control de natalidad y terapia hormonal sustitutiva posmenopáusica100
El embarazo y el parto en pacientes con HAP se asocia con un incremento de la tasa de deterioro y muerte101,102. Aunque se han comunicado casos de embarazos satisfactorios en pacientes con HAPI103, se recomienda la utilización de un método de control de natalidad adecuado en pacientes en edad fértil. Hay consenso entre las guías de la American Heart Association y del American College of Cardiology, en las que se recomienda evitar o interrumpir el embarazo en mujeres con enfermedad cardíaca cianótica congénita, HP y síndrome de Eisenmenger. El Documento de Consenso de Expertos de la ESC sobre el manejo de las enfermedades cardiovasculares durante el embarazo señala que la enfermedad vascular pulmonar severa conlleva una mortalidad materna bien conocida del 30-50%104. De todas formas, no hay consenso entre los expertos acerca del método anticonceptivo más adecuado para estas pacientes. Se cuestiona la seguridad de los anticonceptivos hormonales por su posible efecto protrombótico. Por otra parte, en la actualidad se dispone de una amplia gama de anticonceptivos con dosis bajas de estrógenos y el tratamiento con anticoagulantes orales puede limitar el riesgo de estos agentes. Además, en ensayos recientes con un gran número de pacientes no se ha podido demostrar ninguna relación entre la ingesta de anticonceptivos hormonales y la HAP105. Algunos expertos recomiendan el uso de anticonceptivos libres de estrógenos, la esterilización quirúrgica o los anticonceptivos de barrera. No está claro si el uso de terapia hormonal en mujeres posmenopáusicas con HAP es recomendable o no, pero podría estar indicada en caso de presencia de síntomas menopáusicos intolerables pero asociados con un tratamiento anticoagulante.
Concentraciones de hemoglobina
Los pacientes con HAP son muy sensibles a los descensos en las concentraciones de hemoglobina. Cualquier tipo de anemia leve debe ser tratado sin tardanza. Por otra parte, los pacientes con hipoxia mantenida durante mucho tiempo, como la que se observa en los casos de cortocircuitos derecha-izquierda, tienen tendencia a desarrollar eritrocitosis con concentraciones elevadas de hematocrito. En tales circunstancias están indicadas las flebotomías (véase el apartado Síndrome de Eisenmees > 65% en pacientes sintomáticos (dolor de cabeza, baja concentración) para reducir los efectos adversos de la hiperviscosidad106.
Medicación concomitante
Se extremará el cuidado para evitar una medicación que interfiera con los anticoagulantes orales o que aumente el riesgo de sangrado gastrointestinal. Aunque, según un estudio de casos y controles105, los antiinflamatorios no esteroideos no parecen estar asociados con la HAP, su uso puede reducir aún más la velocidad de filtración glomerular en pacientes con gasto cardíaco bajo y azotemia prerrenal. Los anorexígenos, que han sido asociados con el desarrollo de HAP, ya no están disponibles en el mercado. Se desconoce el efecto de la nueva generación de anorexígenos relacionados con la serotonina, ya que hasta el momento no se han comunicado sus posibles efectos secundarios en el pulmón. Tampoco se ha confirmado la eficacia de los nuevos tratamientos para el fallo cardíaco «biventricular» crónico, como los inhibidores de la enzima de conversión de la angiotensina (IECA) y bloqueadores beta, en pacientes con HAP107. Por otra parte, se desaconseja el uso empírico de estos tratamientos, incluso en dosis bajas, porque pueden desencadenar efectos secundarios severos, como hipotensión y fallo cardíaco derecho.
Asistencia psicológica
Los pacientes con HAP tienen una edad media de alrededor de 40 años y una limitación de la capacidad de ejercicio que puede interferir considerablemente con su estilo de vida previo. Debido a que hoy día se puede obtener información sobre la gravedad y los efectos de la HAP a través de múltiples fuentes, algunas de ellas no profesionales (y, por tanto, pueden no estar actualizadas, ser confusas o demasiado explícitas), muchos pacientes con HAP presentan distintos grados de ansiedad y/o depresión que afectan de forma importante a su calidad de vida. El especialista en HAP debe proporcionar al paciente la información adecuada (informar sobre la gravedad de la enfermedad)108 y referirlo al psicólogo o al psiquiatra cuando lo considere necesario. También son muy útiles los grupos de apoyo para los pacientes y sus familias, coordinados o no por psicólogos o psiquiatras, para una mejor comprensión y aceptación de la enfermedad109.
Cirugía electiva
A pesar de que no hay estudios adecuados sobre este tema, cabe esperar que la cirugía electiva presente un riesgo mayor en los pacientes con HAP. Además, el riesgo podría aumentar según el grado de severidad de la clase funcional de la NYHA y en los casos de intervención abdominal o torácica. No está claro cuál es el tipo de anestesia más recomendable, pero posiblemente la anestesia epidural se tolere mejor que la anestesia general. Esta última debería ser realizada únicamente por anestesistas experimentados con el apoyo de especialistas en HP para decidir el tratamiento más adecuado en caso de complicaciones. Los pacientes en tratamiento con epoprostenol intravenoso y treprostinil subcutáneo deberían tener menos problemas que los pacientes con tratamiento oral o inhalado, ya que éstos pueden resultar afectados por una limitación temporal para la administración de los fármacos, como el ayuno, la anestesia general y la ventilación asistida. En caso de que se prevea la interrupción prolongada del tratamiento (más de 12-24 h) se recomienda cambiar a tratamiento intravenoso para volver al tratamiento original cuando sea posible. El tratamiento anticoagulante se debe interrumpir durante el período más corto posible y se debe iniciar profilaxis para la trombosis venosa profunda.
Tratamiento farmacológico
Tratamiento anticoagulante oral
La utilización del tratamiento anticoagulante oral en pacientes con HAP se basa en la presencia de factores de riesgo tradicionales de tromboembolia venosa, tales como la insuficiencia cardíaca, la vida sedentaria, la demostración de predisposición trombofílica41,42 y de cambios trombóticos en la microcirculación pulmonar5,6 y en las arterias elásticas pulmonares110.
La evidencia de los efectos favorables del tratamiento anticoagulante oral en pacientes con HAPI o HAP asociada con anorexígenos se basa en el análisis retrospectivo de estudios realizados en centros aislados79,111,112. Estos estudios no tenían un diseño aleatorizado y sólo incluían a pacientes con HAPI o con HAP asociada con anorexígenos.
La INR (ratio internacional normalizada) en pacientes con HAPI es de 1,5-2,5 en la mayoría de los centros de Norteamérica y de 2,0-3,0 en los europeos.
La evidencia que apoya la anticoagulación en pacientes con HAPI puede ser extrapolada a otros pacientes con HAP, siempre que se sopese con cuidado la relación riesgo/beneficio.
Por ejemplo, se cree generalmente que el riesgo de sangrado gastrointestinal puede ser más elevado en pacientes con HAP asociada con ETC. Los pacientes con HAP asociada con una enfermedad cardíaca congénita con cortocircuitos intracardíacos presentan mayor riesgo de hemoptisis, pero también pueden tener mayor riesgo de embolia paradójica en la arteria pulmonar y trombosis venosa cerebral27. Los pacientes con hipertensión portopulmonar pueden presentar mayor riesgo de sangrado gastrointestinal debido a la presencia de varices y bajo recuento plaquetario. Los pacientes con HAP en tratamiento con epoprostenol intravenoso indefinido deben estar anticoagulados en ausencia de contraindicaciones, debido en parte al riesgo adicional de trombosis asociada con el cateterismo.
En recientes estudios clínicos aleatorizados se administraron anticoagulantes orales en el 51-86% de los pacientes. Curiosamente, se observó una mayor prevalencia del tratamiento anticoagulante oral en estudios con pacientes con HAPI e insuficiencia cardíaca en clases III y IV de la NYHA, mientras que la prevalencia menor se observó en un estudio en el que sólo se incluía a pacientes con esclerodermia113. Es preciso destacar que no hay evidencia de diferentes niveles de eficacia de la terapia anticoagulante oral según la clase funcional u otros grados de severidad.
Recomendación de clase IIa; nivel de evidencia C para la HAPI. Recomendación de clase IIb; nivel de evidencia C para otras HAP.
Diuréticos
Los pacientes con insuficiencia cardíaca derecha descompensada presentan retención de líquidos que conduce al aumento de la presión venosa central, congestión de órganos abdominales, edema periférico y, en casos avanzados, a la ascitis. En caso de insuficiencia cardíaca derecha, un tratamiento apropiado con diuréticos ofrece beneficios clínicos y sintomáticos claros en pacientes con HAP, aun cuando no se hayan realizado ECA específicos. En recientes ECA sobre nuevos tratamientos, el 49-70% de los pacientes fue tratado con diuréticos; sin embargo, la falta de estudios sobre el tratamiento con clases específicas de diuréticos en la HAP y la variabilidad de las respuestas individuales deja la elección de qué clase de diurético y qué dosis utilizar en casos individuales a la opinión del médico. En los pacientes tratados con diuréticos se debe controlar continuamente los electrolitos séricos y los índices de la función renal.
Recomendación de clase I; nivel de evidencia C.
Oxígeno
La mayoría de los pacientes con HAP (a excepción de los que tienen una enfermedad cardíaca congénita asociada) presentan solamente grados leves de hipoxemia arterial en reposo. En este caso, los mecanismos fisiopatológicos incluyen una baja saturación venosa mixta causada por un gasto cardíaco reducido y tan sólo una mínima discrepancia ventilación-perfusión. En algunos pacientes con hipoxemia profunda podrá documentarse una apertura secundaria del foramen oval permeable. En los pacientes con HAP asociada con defectos cardíacos congénitos, la hipoxemia guarda relación con una inversión del cortocircuito izquierda-derecha, y es refractaria al aumento de oxígeno inspirado.
Por el momento no se dispone de datos consistentes sobre los efectos del tratamiento prolongado con oxígeno en la HAP. Aunque se ha comunicado una mejoría en pacientes con HP tratados con oxígeno suplementario a bajo flujo, ello no ha sido confirmado en estudios controlados. Sin embargo, por lo general se considera importante el mantenimiento de una saturación de oxígeno > 90% en todo momento. El uso del tratamiento con oxígeno en los pacientes con HAP asociada con cortocircuitos cardíacos es más controvertido. De hecho, en un estudio controlado en pacientes con el síndrome de Eisenmenger, la terapia nocturna con oxígeno no tuvo efectos en las variables hemodinámicas, en la calidad de vida o en la supervivencia114. De cualquier modo, se desconoce el efecto de la administración continuada de oxígeno en estos casos.
Recomendación de clase IIa; nivel de evidencia C.
Digital y dobutamina
Debido a que la depresión de la contractilidad del miocardio parece ser uno de los eventos primarios en la progresión del fallo del corazón derecho, se tiene en consideración la administración de agentes inotrópicos para el tratamiento de esta enfermedad. La administración intravenosa a corto plazo de digoxina en los procedimientos de la HAPI provoca un discreto aumento del gasto cardíaco y una reducción significativa de las concentraciones de noradrenalina circulante115. Sin embargo, no hay datos disponibles acerca de los efectos del tratamiento a largo plazo. Por lo tanto, el uso de la digital en pacientes con HAP y fallo refractario del corazón derecho depende básicamente de la opinión del médico, y no de la evidencia científica sobre la eficacia del tratamiento. La digital se puede utilizar en los escasos pacientes con HAP y con fibrilación o aleteo auricular para conseguir una frecuencia ventricular más lenta. En cuanto a la digoxina, se administró en el 18-53% de los pacientes participantes en los ensayos clínicos aleatorizados sobre la HAP más recientes. En la mayoría de los centros especializados en la HAP, los pacientes con HAP terminal son tratados con dobutamina intravenosa116. Este tratamiento normalmente resulta en la mejoría de los síntomas clínicos durante un período variable, como en el caso de insuficiencia cardíaca izquierda avanzada.
Recomendación de clase IIb; nivel de evidencia C.
Bloqueadores de los canales del calcio
La evidencia de la hipertrofia de la media en las arterias pulmonares pequeñas, junto con la reducción de la resistencia vascular pulmonar obtenida mediante fármacos vasodilatadores, llevó a Wood34 hace muchos años a elaborar su hipótesis «vasoconstrictora» como base para la comprensión de la patogenia y fisiopatología de la HAPI. Hoy día se ha demostrado que sólo en una minoría de los pacientes con HAPI, el uso de los vasodilatadores tradicionales como los BCC puede conseguir una reducción significativa de la PAP asociada con un beneficio clínico mantenido a largo plazo.
Los efectos favorables clínicos y pronósticos del tratamiento con altas dosis de BCC en pacientes vasorreactivos con HAPI (véase el apartado Diagnóstico y valoración, para la definición de la respuesta vasorreactiva aguda positiva) han sido demostrados en varios estudios monocéntricos no controlados y no aleatorizados81,79,92,117. En estos estudios, el grupo control estaba formado por pacientes no vasorreactivos que podían tener per se un peor pronóstico que los individuos vasorreactivos92. Sin embargo, no hay evidencia clara para esta hipótesis y parece poco ético negar una terapia con altas dosis de BCC a los pacientes que presentan una reducción importante de la PAP en las pruebas farmacológicas agudas y realizar un estudio con placebo en estos sujetos98.
Los BCC más utilizados en los ensayos clínicos publicados son el nifedipino y el diltiazem, y la elección de uno de ellos puede basarse en la frecuencia cardíaca basal del paciente (una bradicardia relativa favorece la elección de nifedipino y una taquicardia relativa la de diltiazem). Las dosis que han demostrado ser eficaces en la HAPI son relativamente elevadas, hasta 120-240 mg/día de nifedipino y 240-720 mg/día de diltiazem79. En los pacientes vasorreactivos se recomienda comenzar con dosis reducidas (como 30 mg de nifedipino de liberación lenta 2 veces al día o 60 mg de diltiazem 3 veces al día) para ir aumentando con cuidado la dosis de manera progresiva durante las semanas siguientes hasta alcanzar el régimen máximo tolerado. Normalmente, los factores que limitan el incremento de la dosis son la hipotensión sistémica y el edema periférico de las extremidades inferiores. En algunos casos, la adición de digoxina y/o diuréticos puede amortiguar los efectos secundarios de los CBB119. No hay informes acerca de la eficacia, la tolerancia y las dosis efectivas de los CBB de nueva generación, tales como el amlodipino y la flodpina.
Como se ha informado con anterioridad (véase el apartado Diagnóstico y valoración), en general, sólo alrededor del 10-15% de los pacientes con HAPI cumple los criterios para una respuesta vasorreactiva aguda positiva y sólo aproximadamente la mitad de ellos mantendrá a largo plazo una respuesta clínica y hemodinámica positiva al tratamiento con BCC. Por lo general, sólo en estos casos se acepta la continuación del tratamiento único con BCC.
La utilidad del test vasodilatador agudo y del tratamiento indefinido con BCC en pacientes con HAP asociada con la ETC o con la enfermedad cardíaca congénita está menos clara si la comparamos con la HAPI81,86. Sin embargo, los expertos sugieren que incluso en estos casos se realice el test vasodilatador agudo y se trate a los pacientes vasorreactivos con BCC con mucha precaución y estrecha vigilancia para determinar la eficacia y seguridad de dicha terapia.
Los resultados favorables de la administración prolongada de altas dosis de antagonistas de los canales del calcio también han sido demostrados en niños con HAPI118.
Recomendación de clase I; nivel de evidencia C para la HAPI. Recomendación de clase IIb; nivel de evidencia C para otras enfermedades de la HAP.
Prostaciclina sintética y análogos de la prostaciclina
La prostaciclina está producida fundamentalmente por las células endoteliales y es un potente inductor de la vasodilatación de todos los lechos vasculares estudiados. Este componente es el inhibidor endógeno más potente de la agregación plaquetaria y, además, realiza una actividad citoprotectora y antiproliferativa120. En pacientes con HAP se ha mostrado una alteración de las rutas metabólicas de la prostaciclina valorada por una reducción en la expresión de la prostaciclinsintetasa en las arterias pulmonares y de metabolitos urinarios de la prostaciclina13. Aun cuando no sabemos si la alteración de las rutas metabólicas de la prostaciclina tiene un papel causal o es simplemente una consecuencia de la HP, representa un argumento convincente para el uso terapéutico de la prostaciclina en pacientes con HAP. Inicialmente, el uso clínico de la «prostaciclina» (p. ej., epoprostenol) se basaba en sus propiedades vasodilatadoras pulmonares, que fueron demostradas en estudios a corto plazo; este efecto agudo se utiliza actualmente en las pruebas de vasorreactividad de la circulación pulmonar. Por otro lado, incluso los pacientes que no han manifestado una respuesta vasodilatadora aguda al epoprostenol han experimentado una mejoría clínica y hemodinámica con el tratamiento prolongado121. De hecho, la administración prolongada de epoprostenol intravenoso reduce la resistencia vascular pulmonar (RVP) mucho más que los valores que se alcanzan en el test vasodilatador agudo84. La hipótesis que explica estos resultados está basada en los efectos inhibidores de la prostaciclina sobre el crecimiento vascular, el remodelado y la obliteración que podrían facilitar el restablecimiento parcial de las funciones alteradas de la microcirculación pulmonar. Sin embargo, se desconoce el mecanismo de acción exacto de la administración de prostaciclina en la HAP y se cree que puede ser un mecanismo multifactorial; podría incluir la relajación (aguda) de las células musculares lisas, la inhibición y/o normalización de la agregación plaquetaria, la dispersión de los agregados plaquetarios, la mejora de las lesiones de las células endoteliales, la inhibición de la migración y la proliferación de las células vasculares al facilitar el remodelado inverso de los cambios vasculares pulmonares, la mejora del aclaramiento pulmonar de la ET-1, los efectos inotrópicos directos, la mejoría de la utilización periférica del O2 por el músculo esquelético y la mejoría hemodinámica durante el ejercicio13.
El uso clínico de la prostaciclina en los pacientes con HAP se ha extendido por la síntesis de análogos estables que poseen propiedades farmacocinéticas diferentes pero que comparten efectos farmacodinámicos equiparables. Inicialmente, la experiencia en humanos se refiere sobre todo al epoprostenol, una sal sintética de la prostaciclina.
Epoprostenol
El epoprostenol está disponible como preparado liofilizado estable, que ha de ser disuelto en un tampón alcalino (glicina) que permite que dicha solución pueda ser administrada por vía intravenosa. El epoprostenol tiene una vida media corta en la circulación (3-5 min), se convierte rápidamente en fragmentos estables o metabolitos y es estable a temperatura ambiente durante sólo 8 h; ello explica que deba ser administrado por vía intravenosa continua mediante bombas de infusión (p. ej., la bomba CADD®), a través de catéteres permanentes tunelizados (Hickman). El epoprostenol se mantiene a baja temperatura mediante bolsas refrigerantes, que permiten reemplazar la infusión diariamente. No se recomienda el uso de catéteres subcutáneos con reservorios y agujas transcutáneas (utilizadas en tratamientos intermitentes).
La eficacia de la administración continua por vía intravenosa del epoprostenol (prostaciclina sintética) ha sido probada en 3 estudios clínicos controlados abiertos sobre la HAPI121,122 y sobre la HAP asociada con el espectro de enfermedades de la esclerodermia113 (tabla 12). El epoprostenol mejora los síntomas, la capacidad de ejercicio y la hemodinámica en ambas enfermedades y es el único tratamiento que mejora la supervivencia en la HAPI, como se ha probado en ensayos clínicos aleatorizados.

Recientemente se han comunicado los datos relativos a 2 grandes series de pacientes con HAPI tratados con epoprostenol77,87. Los datos muestran una supervivencia de alrededor del 65% a los 3 años, que fue relacionada con la severidad basal de la enfermedad y la respuesta a la terapia a los 3 meses. Los autores sugirieron la necesidad del trasplante de pulmón para el subgrupo de pacientes que permanecían en la clase funcional III o IV de la NYHA, o para los que no alcanzaron una mejoría hemodinámica o de la capacidad de ejercicio significativa a los 3 meses de tratamiento con epoprostenol, o ambos.
El tratamiento prolongado con epoprostenol se inicia con una dosis de 2-4 ng/kg/min y se incrementa gradualmente dependiendo de los efectos secundarios (rubor, dolor de cabeza, diarrea, dolor de miembros inferiores). La dosis que se debe alcanzar durante las primeras 2-4 semanas es, normalmente, de alrededor de 10-15 ng/kg/min; entonces es preciso el incremento periódico de la dosis para maximizar la eficacia y mantener los resultados debido a la posible tolerancia del fármaco. La dosis óptima varía según los individuos y en la mayoría de los casos se sitúa entre 20 y 40 ng/kg/min, aunque la estrategia de aumento de la dosis es diferente según el centro. En 2 grandes series de pacientes tratados con epoprostenol77,87 publicadas recientemente, la dosis media fue de 21 ± 7 y 27 ± 8 ng/kg/min, respectivamente.
Los efectos adversos del tratamiento indefinido con epoprostenol son muy comunes y entre ellos se incluyen rubor, dolor o malestar en distintas partes del cuerpo (mandíbula, cabeza, espalda, pies y piernas), cólico abdominal, náuseas y rara vez hipotensión. La incidencia de los efectos secundarios puede depender de la agresividad con que se aumente la dosis inicialmente. Sólo es preciso reducir la dosis si la intensidad es moderada o severa. Puede haber recurrencia de los efectos secundarios tras el incremento de la dosis, aunque normalmente son leves y autolimitados en el tiempo sin cambios en la dosis. Se han observado algunos casos de ascitis que podrían estar relacionados con un aumento de la permeabilidad de la membrana peritoneal inducida por el epoprostenol. Los eventos adversos relacionados con el sistema de administración del fármaco son más serios y se suelen relacionar con el mal funcionamiento de la bomba, infección local, obstrucción del catéter y sepsis. En 2 grandes series77,87 se informó que 0,14 y 0,19 episodios de sepsis por paciente/año y 8 muertes (2,8%) de un total de 340 pacientes estuvieron directamente relacionadas con infecciones del catéter. También pueden ocurrir infecciones localizadas, tales como reacciones a las zonas pequeñas de salida, infecciones de los túneles y celulitis. Los eventos raros son el neumotórax y el hemotórax, que pueden ocurrir durante la inserción del catéter. Se debe evitar la interrupción brusca de la infusión de epoprostenol debido a que en algunos pacientes esto puede provocar un empeoramiento de la HP como efecto rebote, con deterioro sintomático e incluso muerte. El manejo de los pacientes con tratamiento indefinido con epoprostenol requiere una infraestructura considerable, así como personal médico y de enfermería experimentado.
A pesar de que los ensayos clínicos aleatorizados han sido realizados solamente en la HAPI y en la HAP asociada con la esclerodermia, se han observado resultados favorables en estudios no controlados en otros subgrupos, como en la HAPI pediátrica118, el lupus sistémico eritematoso123 y otras enfermedades del tejido conectivo124, en la HAP asociada con defectos cardíacos congénitos con cortocircuitos sistémico-pulmonares, reparados o no124,125, en la hipertensión portopulmonar124,126, la HAP asociada con la enfermedad de Gaucher127 y en la infección por VIH128. No hay consenso entre los especialistas sobre la efectividad del tratamiento con epoprostenol en pacientes con HP asociada a tromboembolia crónica no operable, aunque se han observado algunos efectos positivos129.
En Europa, el epoprostenol no ha sido registrado a través del procedimiento centralizado de la Unión Europea (Agencia Europea para la Evaluación de Productos Médicos [EMEA]), pero en algunos países está aprobado para la HAPI en clase funcional III y IV de la NYHA. En Estados Unidos y Canadá, la Food and Drug Administration (FDA) ha aprobado la utilización del epoprostenol para la HAPI y la HAP asociada con ETC en clase funcional III y IV de la NYHA.
Recomendación de clase I; nivel de evidencia A para la HAPI y la HAP asociada con ETC. Recomendación de clase IIa; nivel de evidencia C para otras enfermedades de la HAP.
Se han realizado 4 ensayos clínicos aleatorizados con análogos de la prostaciclina que aparecen resumidos en la tabla 13.

Treprostinil
El treprostinil es un bencideno tricíclico análogo del epoprostenol, con suficiente estabilidad química para ser administrado a temperatura ambiente diluido en una solución fisiológica. Estas características permiten la administración del compuesto por vía intravenosa y también subcutánea. La administración subcutánea del treprostinil se puede realizar mediante bombas de microinfusión (Mini-Med®) y pequeños catéteres subcutáneos similares a los utilizados para la administración de insulina en pacientes diabéticos. En este caso, se pueden evitar los problemas relacionados con la vía venosa central (como infecciones) y el manejo del sistema es mucho más sencillo.
Los efectos de la administración subcutánea continuada del treprostinil en la HAP han sido estudiados en el mayor ensayo clínico internacional aleatorizado realizado hasta el momento, en el que se observó una mejoría de la capacidad de ejercicio y de los eventos hemodinámicos y clínicos130. El mayor aumento de la capacidad de ejercicio se observó en pacientes con mayor compromiso en estado basal y en pacientes que no toleraban la dosis del cuartil superior (dosis > 13,8 ng/kg/min). En un primer estudio piloto controlado realizado con treprostinil en 26 pacientes se observó una tendencia a la mejoría en la prueba de la marcha de 6 min y en la reducción de la RVP131.
El efecto secundario más común fue el dolor en la zona de infusión, que llevó a la interrupción del tratamiento en el 8% de los casos con fármaco activo y a la limitación del incremento de la dosis en otro porcentaje adicional de pacientes. La mortalidad total fue del 3% y no se observaron diferencias entre los grupos de tratamiento. En los informes preliminares se apuntó la posibilidad de la transferencia de pacientes en tratamiento con epoprostenol intravenoso a tratamiento con treprostinil subcutáneo132.
En 2002, la FDA aprobó el uso del treprostinil en pacientes con HAP en clase funcional II, III y IV.
Recomendación de clase IIa; nivel de evidencia B para la HAP.
Beraprost sódico
El beraprost sódico es el primer análogo de la prostaciclina químicamente estable y oralmente activo. Se absorbe con rapidez cuando se toma en ayunas, la concentración pico se alcanza a los 30 min y la vida media de eliminación es de 35-40 min después de una administración oral única.
El beraprost (un análogo de la prostaciclina oralmente activo) ha sido evaluado en pacientes con HAP en 2 ensayos clínicos aleatorizados realizados en Europa133 y en Estados Unidos74 (tabla 2). En el primer estudio, el fármaco se administró oralmente 4 veces al día con la dosis tolerada más alta (dosis media de 80 mg 4 veces al día); el aumento de la capacidad de ejercicio se observó solamente en los pacientes con HAPI después de 3 meses. En el segundo estudio aleatorizado, que se desarrolló durante 12 meses, la mejora en la capacidad de ejercicio se observó a los 3 y 6 meses, pero no se mantuvo después de este período. No se observó mejoría hemodinámica en el estudio a largo plazo y sólo se apreció una reducción de los eventos clínicos en el seguimiento a los 6 meses.
El beraprost sódico ha sido aprobado en Japón y Corea del Sur para la HAPI, pero en Europa y Estados Unidos su aprobación parece que ha sido paralizada.
Recomendación de clase IIb; nivel de evidencia B para la HAPI.
Iloprost inhalado
El iloprost es un análogo de la prostaciclina químicamente estable, preparado para su administración intravenosa, oral e inhalada. La terapia inhalada es muy atractiva y en teoría cuenta con la ventaja de ser selectiva para la circulación pulmonar. De hecho, debido a que las arterias intraacinares pulmonares están estrechamente rodeadas de unidades alveolares, se puede dilatar estos vasos mediante la deposición alveolar del vasodilatador. Es fundamental que las partículas inhaladas sean lo suficientemente pequeñas (3-5 µm de diámetro) para asegurar la deposición alveolar.
Tras la inhalación de iloprost se observó una reducción del 10-20% de la PAP media, que se mantuvo durante 45-60 min134. La corta duración de su acción hace necesarias frecuentes inhalaciones (de 6 a 12 veces al día) para obtener un efecto persistente con la administración a largo plazo. Con los nebulizadores a presión (Jet), cada inhalación dura alrededor de 15 min; otros dispositivos alternativos (como los nebulizadores ultrasónicos) permiten reducir el período de inhalación a unos 5 min.
En un ensayo clínico aleatorizado se evaluó el iloprost inhalado (6-9 veces; 2,5-5 μg/inhalación, 30 μg de media diaria) comparado con la inhalación de placebo en pacientes con HAP y HPTEC135 (tabla 13). El estudio mostró un aumento de la capacidad de ejercicio y una mejora de los síntomas, la RVP y los eventos clínicos solamente en pacientes con HAPI. En general, el iloprost inhalado fue bien tolerado: la presencia de tos fue más frecuente en el grupo de tratamiento con iloprost, así como el rubor y las cefaleas.
Se han comunicado los resultados de un estudio a largo plazo, no controlado, en 25 pacientes con HAPI tratados durante al menos 1 año con 100-150 mg diarios de iloprost inhalado136; los datos mostraron un aumento medio de 85 m en la prueba de la marcha de 6 min, una reducción de 7 mmHg en la PAP media y un aumento del índice cardíaco de 0,6 l/min/m². En un pequeño estudio realizado en 8 pacientes con HP y fibrosis pulmonar, la administración aguda de iloprost inhalado provocó una marcada vasodilatación pulmonar, con mantenimiento del intercambio gaseoso y de la presión arterial sistémica137, lo que apunta a la posible utilidad del tratamiento en este subgrupo particular de pacientes.
El tratamiento con iloprost inhalado ha sido aprobado en Europa por la EMEA para la HAPI en clase III de la HYHA y en Australia y Nueva Zelanda para la HAP y la HPTEC no operable en clase III y IV.
Recomendación de clase IIa; nivel de evidencia B para la HAPI.
Iloprost intravenoso
La administración intravenosa continua de iloprost parece ser tan efectiva como el epoprostenol, según los resultados de algunos estudios pequeños en pacientes con HAP y HPTEC138,139. El iloprost tiene la ventaja de ser estable a temperatura ambiente y no necesita ser reconstituido o refrigerado.
La administración intravenosa continua de iloprost ha sido aprobada en Nueva Zelanda para la HAP y clase funcional III y IV de la NYHA.
Recomendación de clase IIa; nivel de evidencia C para la HAP.
Antagonistas de los receptores de la endotelina 1
La ET-1, un péptido producido fundamentalmente por células endoteliales vasculares, se caracteriza por su poder vasoconstrictor y mitógeno para el músculo liso14. La ET-1 se une a 2 tipos de receptores, ETA y ETB: los receptores ETA se encuentran en las células musculares lisas, mientras que los receptores ETB se localizan tanto en las células endoteliales como en las células musculares lisas. La activación de los receptores ETA y ETB de las células musculares lisas interviene en los efectos vasoconstrictores y mitógenos de la ET-1. La estimulación de los receptores endoteliales ETB promueve el aclaramiento de la ET-1, la activación del NO y la producción de prostaciclina.
La activación del sistema de la ET-1 se ha observado tanto en el plasma140 como en el tejido pulmonar de los pacientes con HAP141. Aunque no se ha determinado si el aumento de las concentraciones de ET-1 en el plasma es una causa o una consecuencia de la HP140, los estudios sobre la expresión tisular del sistema de la ET sugieren que la ET-1 tiene un papel importante en la patogenia de la HAP14.
La evidencia indiscutible de la activación del sistema de la ET en la HAP constituye un argumento de peso para estudiar los efectos de los antagonistas de la ET-1 en los pacientes con HAP. La forma más eficiente de antagonizar el sistema de la ET-1 es utilizando los antagonistas de los receptores de la ET-1, ya que éstos pueden bloquear tanto a los receptores ETA como a ambos receptores, ETA y ETB.
En la actualidad se están desarrollando 3 ensayos clínicos aleatorizados con antagonistas de los receptores de la ET-1 en pacientes con HAP (tabla 14).

Bosentán
El bosentán es un antagonista oral, dual y activo de los receptores ETA y ETB y es la primera molécula de esta clase de fármacos que ha sido sintetizada142. El efecto del bosentán ha sido estudiado en la HAP en 2 ensayos clínicos aleatorizados que han mostrado una mejoría en la capacidad de ejercicio, en la clase funcional, en la hemodinámica, en las variables ecocardiográficas y Doppler y en la evolución de la enfermedad58,143,144. En el estudio BREATHE-1, los pacientes fueron aleatorizados en 3 grupos (1:1:1) para recibir placebo o 62,5 mg de bosentán 2 veces al día durante 4 semanas, seguido de 125 o 250 mg de bosentán 2 veces al día, durante un período mínimo de 12 semanas. Aunque ambas dosis de bosentán indujeron efectos significativos del tratamiento, se observó una mejoría (corregida respecto al placebo) más pronunciada en el grupo con la dosis de 250 mg frente al de 125 mg (+54 y +35 m de efecto del tratamiento en la prueba de la marcha de 6 min, respectivamente). Sin embargo, no se pudo determinar la respuesta formal a la dosis para la eficacia. Por otra parte, si bien se observó un efecto similar del tratamiento en los pacientes con HAPI y en los pacientes con HAP asociada con esclerodermia, el bosentán mejoró la distancia caminada, comparada con la distancia basal, en los pacientes con HAPI (+46 m en el grupo de bosentán frente a -5 m en el grupo placebo), mientras que solamente previno el deterioro de la distancia caminada en los pacientes con esclerodermia (+3 m en el grupo de bosentán frente a -40 m en el grupo placebo). Se produjo elevación de las aminotransferasas hepáticas en el 10% de los sujetos, que demostraron ser dependientes de la dosis y reversibles tras la reducción o interrupción de la dosis. De hecho, las anomalías de la función hepática fueron más frecuentes y severas en el grupo tratado con 250 mg de bosentán y, en todos los casos en los que se disminuyó la dosis del fármaco, se observó una reducción de las concentraciones de transaminasas. Sobre la base de estos resultados, se confirmó la dosis de 125 mg de bosentán 2 veces al día como la dosis terapéutica recomendada. El mecanismo más probable de los cambios enzimáticos hepáticos con el tratamiento con bosentán es la competición (dosis-dependiente) entre el bosentán y sus metabolitos y la excreción de sales biliares que son retenidas y pueden ser citotóxicas para los hepatocitos145.
En una ampliación de este estudio se administró bosentán a 29 pacientes: los pacientes evaluados en el seguimiento a los 6 meses mostraron una mejoría en la distancia caminada y el tratamiento prolongado con bosentán (> 1 año) se asoció con la mejoría de los parámetros hemodinámicos y la clase funcional de la NYHA146.
Recientemente, el bosentán oral ha sido propuesto como terapia de transición para pacientes que mostraron efectos secundarios severos y/o con intolerancia a la terapia con prostanoides, incluida la sepsis con epoprostenol intravenoso147.
El estudio BREATHE-3 (un estudio no controlado, abierto, monocéntrico y de dosis múltiple), realizado en niños de 4-17 años de edad, fue diseñado para evaluar las propiedades fármacocinéticas, la tolerancia y la seguridad del bosentán oral. En este estudio preliminar se observó una mejoría significativa de los parámetros hemodinámicos después de 12 semanas de tratamiento en los 18 niños tratados con bosentán solo o combinado con epoprostenol148.
Debido al potencial incremento de enzimas hepáticas, la FDA exige la realización de pruebas de la función hepática al menos 1 vez al mes. La EMEA recomienda el control mensual de las pruebas hepáticas; estos datos están siendo recogidos en una base de datos accesible en Internet (TRAX). Es recomendable también vigilar regularmente la hemoglobina y los hematocritos, ya que el uso de bosentán puede estar asociado con el desarrollo de anemia, aunque ésta suele ser leve. Además, se ha observado retención de líquidos y edema de las extremidades inferiores en pacientes tratados con bosentán. Se debe prestar especial atención al uso de anticonceptivos en las mujeres en edad fértil, debido al potencial efecto teratogénico del bosentán. Este fármaco puede disminuir la eficacia de los anticonceptivos hormonales y por esta razón se debe buscar una mayor protección. Hay la preocupación de que los antagonistas de la endotelina como clase puedan causar atrofia testicular e infertilidad del varón, por lo que los varones en edad fértil deben ser aconsejados con respecto a este tema antes de tomar la medicación.
El bosentán ha sido aprobado para el tratamiento de los pacientes con HAP en clase funcional III y IV de la NYHA en Estados Unidos y Canadá. En Europa, la EMEA ha aprobado el bosentán para el tratamiento de pacientes en clase funcional III de la NYHA, especificando que la eficacia del tratamiento sólo ha sido demostrada en pacientes con HAPI y con HAP asociada con esclerodermia sin fibrosis pulmonar significativa.
Recomendación de clase I; nivel de evidencia A para la HAPI en clase funcional III de la NYHA y para la HAP asociada a esclerodermia sin fibrosis pulmonar significativa.
Recomendación de clase IIa; nivel de evidencia B para la HAPI en clase funcional IV de la NYHA y para la HAP asociada a esclerodermia sin fibrosis pulmonar significativa.
Sitaxsentan
El uso del sitaxsentan, un antagonista selectivo y oralmente activo del receptor ETA, ha sido evaluado en pacientes con HAP en un ensayo clínico aleatorizado en el que se incluyó a 178 pacientes con HAP en clase funcional II, III y IV de la NYHA75. La etiología incluía la HAPI y la HAP asociadas con ETC o con enfermedades cardíacas congénitas. Los pacientes fueron aleatorizados en 1:1:1 para recibir placebo, 100 mg de sitaxsentan o 300 mg de sitaxsentan oral 1 vez al día durante 12 semanas. El estudio demostró una mejoría en la capacidad de ejercicio, la hemodinámica y los eventos clínicos75. La incidencia de anomalías en las pruebas de la función hepática, que revirtieron en todos los casos, fue del 0% para 100 mg y del 9,5% para 300 mg. En un estudio piloto adicional con este componente realizado en 20 pacientes con HAP se obtuvieron resultados similares149.
El sitaxsentan puede aumentar la INR o el tiempo de protrombina debido a la inhibición de la enzima CYP2C9 P450, la principal enzima hepática que interviene en el metabolismo de la warfarina (cumarínicos). Esta interacción se puede controlar reduciendo la dosis de warfarina para alcanzar la INR deseada.
En la actualidad se está desarrollando un segundo estudio clínico con sitaxsentan para evaluar más a fondo la eficacia y los efectos secundarios, así como para obtener la aprobación de las autoridades competentes. En la presente guía no se establece el grado de recomendación porque el sitaxsentan sólo está disponible para pacientes que participan en ensayos clínicos.
Recomendación de clase no determinada en la actualidad; nivel de evidencia B.
Ambrisentan
La utilización del ambrisentan, un antagonista selectivo y oralmente activo del receptor ETA, ha sido evaluada en un estudio piloto ciego de comparación de dosis en 64 pacientes con HAP. Los resultados preliminares muestran una mejoría en la capacidad de ejercicio y en la hemodinámica que parece similar a las obtenidas con otros antagonistas de los receptores de la endotelina (ARE)150. En la actualidad se están desarrollando 2 estudios clínicos aleatorizados con ambrisentan para evaluar más a fondo la eficacia y los efectos secundarios y obtener la aprobación de las autoridades competentes.
No establecemos el grado de recomendación porque el ambrisentan sólo está disponible para pacientes participantes en ensayos clínicos.
Recomendación de clase no determinada en la actualidad; nivel de evidencia C.
Inhibidores de la fosfodiesterasa tipo 5
Sildenafilo
El sildenafilo es un potente y selectivo inhibidor de la GMP cíclica de la fosfodiesterasa tipo 5 (FDE-5) oralmente activo, que ejerce su efecto farmacológico por el aumento de la concentración intracelular de la cGMP151. El aumento de este nucleótido induce efectos de relajación y antiproliferación en las células musculares lisas vasculares152. La FDE-5 es abundante de forma selectiva en la circulación pulmonar153,154 y la expresión y actividad del gen de la FDE-5 aparecen aumentadas en la HP crónica155,156. Esto sugiere que el sildenafilo podría tener un efecto preferente en el árbol vascular pulmonar.
En varios ensayos clínicos no controlados se han comunicado los efectos favorables del sildenafilo (inhibidor oralmente activo de la FDE-5) en la HAP157-159, en la HPTEC160 y en la HP asociada con fibrosis pulmonar161. Este fármaco, en dosis de 25-75 mg 3 veces al día, parece mejorar tanto la hemodinámica cardiopulmonar como la capacidad de ejercicio. En dichos estudios se observaron también relativamente pocos efectos secundarios leves (cefalea, congestión nasal y alteraciones en la vista).
Recientemente se han publicado los resultados de un estudio aleatorizado con diseño cruzado: la administración de 25-100 mg de sildenafilo 3 veces al día en 22 pacientes con HAP en clase II y III de la NYHA mejoró los síntomas de la enfermedad al cabo de 6 semanas, así como la capacidad de ejercicio que fue evaluada mediante el protocolo Naughton en la cinta de ejercicio (de 475 ± 168 s de ejercicio al final de la fase placebo a 686 ± 224 s al final de la fase con sildenafilo) y los parámetros hemodinámicos15. Los resultados de un importante ensayo clínico aleatorizado realizado en 278 pacientes con HAP en clase II y III de la NYHA fueron presentados en el congreso de la American College of Chest Physicians a finales de octubre de 2004. Los datos mostraron que los efectos del tratamiento (corregido por placebo) en la prueba de la marcha de 6 min fueron de alrededor de 45 m para dosis de 20, 40 y 80 mg de sildenafilo 3 veces al día. Todos los rangos de dosis redujeron la PAP media en la semana 12 de tratamiento entre 3 y 5 mmHg. En el momento de publicación de esta guía, el uso del sildenafilo todavía no ha sido aprobado por ningún organismo competente para el tratamiento de la HAP162. En este momento se debe considerar el tratamiento con sildenafilo en pacientes con HAP en los que las terapias aprobadas no han surtido efecto o que no son candidatos para ellas.
Recomendación de clase I; nivel de evidencia A.
Terapia combinada
La terapia combinada es una opción atractiva para el manejo de los múltiples mecanismos fisiopatológicos que están presentes en la HAP. La terapia combinada consiste en la iniciación simultánea de 2 (o más) tratamientos o en la adición de un segundo (o tercer) tratamiento a una terapia previa si ésta se considera insuficiente. Por el momento se desconoce cuál de estas 2 estrategias es la más adecuada.
La eficacia y seguridad de la iniciación concurrente de bosentán y epoprostenol fueron investigadas en un ensayo clínico (BREATHE-2) que incluía a 33 pacientes con HAP en clase III y IV de la NYHA. Los pacientes fueron asignados de forma aleatoria al grupo de tratamiento con epoprostenol + placebo o al grupo de epoprostenol + bosentán. En ambos grupos se observó una mejoría de los parámetros hemodinámicas, la capacidad de ejercicio y la clase funcional. Los datos muestran una tendencia hacia una mejora mayor (aunque no significativa) en los parámetros hemodinámicos en el grupo de epoprostenol + bosentán163. Sin embargo, también se observó un aumento de los eventos adversos en el grupo de terapia combinada comparado con el grupo con tratamiento con epoprostenol solo.
En este momento se están desarrollando o programando nuevos ensayos clínicos para estudiar los efectos de la adición de sildenafilo a los pacientes que reciben tratamiento con epoprostenol.
En los pacientes con HAP que presentaban deterioro a pesar del tratamiento crónico con prostanoides no parenterales, la adición de bosentán164 o sildenafilo165 al tratamiento instaurado tuvo efectos favorables en la hemodinámica pulmonar y en la capacidad de ejercicio. Estos datos fueron observados en ensayos clínicos no controlados (*).
(*) Recientemente1 se ha observado en pacientes con hipertensión arterial pulmonar tratados crónicamente con prostanoides (parenterales y no parenterales) que habían iniciado deterioro clínico, incremento en su capacidad funcional, remodelado favorable del ventrículo derecho y disminución de los episodios de insufiencia cardíaca derecha descompensada al añadir sildenafilo al tratamiento.
1. Jiménez López-Guarch C, Escribano Subias P, Tello de Meneses R, Delgado Jiménez J, Sadia Pérez D, Velázquez Martín T, et al. Eficacia del sildenafilo por vía oral como terapia de rescate en pacientes con hipertensión arterial pulmonar severa en tratamiento crónico con prostaciclina. Resultados a largo plazo. Rev Esp Cardiol. 2004;57:946-51.
Recomendación de clase IIb; nivel de evidencia C.
Procedimientos intervencionistas
Septostomía auricular con balón
Varias observaciones experimentales166 y clínicas167 apuntan a que la presencia de una comunicación interauricular (CIA) podría ser beneficiosa en caso de HP severa. De hecho, la presencia de una CIA permitiría que los cortocircuitos derecha-izquierda aumentaran el gasto sistémico que, a pesar del descenso en la saturación de oxígeno arterial sistémico, produciría un aumento del transporte de oxígeno. Además, en la aurícula, el cortocircuito permitiría la descompresión de la aurícula y el ventrículo derechos, aliviando los signos y síntomas del fallo del corazón derecho.
No se ha establecido el papel de la septostomía auricular con balón en el tratamiento de los pacientes con HAP debido a que su eficacia sólo se conoce mediante estudios en pequeñas series de pacientes y casos comunicados, que suman un total de alrededor de 120 pacientes168,169. En la mayoría de los casos, esta intervención fue realizada en pacientes gravemente enfermos como terapia paliativa y de puente para el trasplante de pulmón, factores que podrían explicar una tasa de mortalidad del procedimiento del 5-15%. Sin embargo, se ha observado una mejora sintomática y hemodinámica, además de un aumento en la supervivencia, al compararlos con grupos históricos de control76. En el presente, la septostomía auricular con balón está indicada en pacientes con clase funcional III y IV de la NYHA, con síncope recurrente y/o fallo del corazón derecho, que no responden a los tratamientos médicos disponibles; la septostomía se utiliza como terapia paliativa y de puente para el trasplante pulmonar, o como tratamiento único cuando no se dispone de otras opciones169. La septostomía auricular con balón sólo debe ser realizada en centros con amplia experiencia para reducir los riesgos del procedimiento.
Recomendación de clase IIa; nivel de evidencia C.
Trasplante de pulmón
El trasplante de pulmón y de pulmón-corazón en la HAP sólo ha sido estudiado en series prospectivas no controladas, debido a que no se considera ético el diseño de estudios aleatorizados formales cuando no hay otras alternativas de tratamiento169.
La supervivencia a los 3 y 5 años tras el trasplante de pulmón y de pulmón-corazón es de aproximadamente el 55 y el 45%, respectivamente170.
En la HAPI se ha realizado tanto el trasplante pulmonar unilateral como bilateral y, en algunos casos, estas operaciones se combinaron con la reparación de defectos cardíacos del síndrome de Eisenmenger. Las tasas de supervivencia de los receptores fueron similares en los trasplantes unilateral y bilateral y, siempre que sea técnicamente factible, cualquiera de estas 2 operaciones es una alternativa aceptable en la mayoría de los casos de HAP. Sin embargo, en muchos centros de trasplante actualmente se prefiere el trasplante pulmonar bilateral porque, por lo general, presenta menos complicaciones postoperatorias. En los pacientes con el síndrome de Eisenmenger y en los que presentan insuficiencia cardíaca en fase terminal, la opción del trasplante de corazón y pulmón debe ser estudiada con especial atención. En algunos casos de defectos complejos y en casos de comunicación interventricular se ha observado una mayor supervivencia en el trasplante de corazón y pulmón.
El trasplante de pulmón y de pulmón-corazón está indicado en pacientes con HAP sintomáticos en clase III avanzada y IV de la NYHA, refractarios a los tratamientos médicos disponibles. La imposibilidad de predicción del tiempo de espera y la escasez de donantes de órganos complican la toma de decisiones en relación con el momento apropiado para la inclusión en la lista de trasplante.
Recomendación de clase I; nivel de evidencia C.
Algoritmo de tratamiento
En la figura 3 se expone el algoritmo de tratamiento que ha sido elaborado según el grado de recomendación y el nivel de evidencia.

Fig. 3. Algoritmo de tratamiento basado en la evidencia. (1) El algoritmo está restringido a pacientes con clase funcional III o IV de la NYHA ya que éstos representan la mayor población de pacientes incluidos en ensayos clínicos controlados. Se dispone de muy pocos datos sobre la clase I y II de la NYHA. Además, los distintos tratamientos han sido evaluados fundamentalmente en pacientes con hipertensión arterial pulmonar idiopática (HAPI) esporádica y en la HAP asociada con esclerodermia o con la utilización de anorexígenos. La extrapolación de estas recomendaciones a otros subgrupos de la HAP se debe realizar con precaución. (2) Debido a la complejidad del test vasodilatador agudo y de las opciones disponibles de tratamiento, se recomienda encarecidamente que se tome en consideración referir al paciente con HAP a un centro especializado. (3) Todos los pacientes con HAP deben ser sometidos al test vasodilatador agudo, a pesar de que el mayor número de respuestas positivas se observa en los pacientes con HAPI y HAP asociada al uso de anorexígenos. (4) La respuesta aguda positiva a los vasodilatadores se define como un descenso de la presión arterial pulmonar media de al menos 10 mmHg, hasta ≤ 40 mmHg, con un gasto cardíaco aumentado o sin cambios durante la fase aguda con NO inhalado, epoprostenol i.v. o adenosina i.v. (5) La respuesta sostenida a los bloqueadores de los canales de calcio (BCC) se define como la presencia de una hemodinámica casi normal en pacientes con clase funcional I o II de la NYHA después de varios meses de tratamiento. (6) En los pacientes con clase funcional III de la NYHA, la terapia de primera línea puede incluir antagonistas orales de los receptores de la endotelina, epoprostenol i.v. indefinido o análogos prostanoides. (7) En el momento de la publicación de esta guía, el sildenafilo no ha sido aprobado para la HAP por ningún organismo regulador. (8) La mayoría de los expertos consideran que los pacientes con clase funcional IV de la NYHA en condiciones inestables deben ser tratados con epoprostenol i.v. (mejoría de la supervivencia, experiencia en todo el mundo y acción rápida). Los grados A, B, C de acuerdo a las tablas 4 y 5; BCC: bloqueadores de los canales del calcio; inh: inhalado; i.v.: intravenoso continuo; PDE: fosfodiesterasa; R: receptores.
El algoritmo está restringido a pacientes en clase funcional III o IV de la NYHA, ya que éstos representan la población predominante incluida en los ensayos clínicos aleatorizados.
En relación con los pacientes en clase I o II de la NYHA, hay muy pocos datos y la estrategia de tratamiento más adecuada todavía está por determinar e incluso validar mediante ensayos específicos. Por el momento, estos pacientes deber ser tratados con terapia de mantenimiento y, en caso de vasorreactividad, con BCC. En casos en los que haya múltiples indicadores de pronóstico favorable (véase el apartado Valoración de la severidad) se recomienda aplicar una estrategia de vigilancia y espera o incluir al paciente en un ensayo clínico aleatorizado.
Los distintos tratamientos han sido evaluados fundamentalmente para la HAPI y para la HAP asociada con esclerodermia o uso de anorexígenos. La extrapolación de estas recomendaciones a otros subgrupos de la HAP debe realizarse con mucho cuidado (véase el apartado Enfermedades específicas).La estrategia inicial que ha sido sugerida, tras el establecimiento del diagnóstico de HAP, consiste en la adopción de las medidas generales y la iniciación de la terapia de mantenimiento que incluye la anticoagulación oral (si no está contraindicada), diuréticos en caso de retención de líquidos, oxígeno suplementario en caso de hipoxemia y digoxina en caso de fallo refractario del corazón derecho y/o arritmias supraventriculares.
Debido a la complejidad de los estudios adicionales y de los tratamientos disponibles, se recomienda encarecidamente que los pacientes con HAP sean referidos a centros especializados.
El test vasodilatador agudo se debe realizar en todos los pacientes con HAP, aunque los pacientes con HAPI asociada con el uso de anorexígenos son los que tienen más probabilidades de presentar una respuesta aguda positiva y los que se beneficiarán de la terapia con altas dosis de BCC.
Los pacientes vasorreactivos, como se define con anterioridad, deben ser tratados con las dosis de CBB de óptima tolerancia; la respuesta mantenida (definida como clase funcional I o II de la NYHA con hemodinámica prácticamente normal) debe ser confirmada después de 3-6 meses de tratamiento.
Los pacientes que no responden al test vasodilatador agudo y que se encuentran en clase funcional I y II de la NYHA deben continuar con la terapia de mantenimiento bajo estrecha vigilancia médica.
Los pacientes que no responden al test vasodilatador agudo o que responden pero permanecen en clase funcional III de la NYHA deben ser considerados candidatos para el tratamiento con antagonistas de los receptores de la endotelina (ARE) o con prostanoides. Por el momento, el único ERA aprobado y disponible en el mercado es el bosentán, un antagonista dual y oralmente activo. Entre los prostanoides, el treprostinil se administra por vía subcutánea y ha sido aprobado en Estados Unidos; el iloprost inhalado está aprobado en Europa y Australia, mientras que el beraprost lo está en Japón y Corea del Sur. La administración continua de epoprostenol intravenoso puede ser utilizada en pacientes en clase III de la NYHA que son refractarios a los ARE u otros prostanoides. Algunos autores prefieren utilizar epoprostenol como primera elección en pacientes en clase III de la NYHA sobre la base de los beneficios demostrados de este fármaco en la supervivencia.
La elección del fármaco depende de una serie de factores, entre los que se incluye el estado de aprobación, la vía de administración, el perfil de los efectos secundarios, las preferencias del paciente y la experiencia del médico.
El sildenafilo, un inhibidor oralmente activo de la fosfodiesterasa-5, todavía no ha sido aprobado por ningún organismo competente y su uso puede ser considerado en sujetos que no responden a otras terapias o que no son candidatos para éstas. El papel de este fármaco quedará mejor establecido cuando las autoridades competentes evalúen los datos de un importante estudio clínico aleatorizado realizado con este agente.
La administración continua de epoprostenol intravenoso (aprobado en Europa y Estados Unidos) puede ser considerada la terapia de primera elección en pacientes con HAPI en clase funcional IV de la NYHA, sobre la base de los beneficios demostrados en la supervivencia de este subgrupo.
A pesar de que tanto el bosentán como el treprostinil han sido aprobados para los pacientes en clase funcional IV, sólo un pequeño número de estos pacientes fue incluido en los ensayos clínicos realizados con estos agentes. En consecuencia, la mayoría de los expertos considera que son tratamientos de segunda elección para pacientes gravemente enfermos. Aunque el iloprost intravenoso no ha sido estudiado en ensayos clínicos aleatorizados, el uso de este análogo de la prostaciclina ha sido aprobado en Nueva Zelanda.
La terapia combinada (ARE + prostanoides) se puede tener en cuenta en pacientes que no presentan mejoría o con deterioro con el tratamiento de primera elección, aunque los datos sobre esta estrategia específica son limitados y, por el momento, todavía no son controlados. Es preciso implementar protocolos adecuados de planificación y dosificación de la terapia combinada para limitar los posibles efectos secundarios.
La septostomía auricular con balón y/o el trasplante de pulmón están indicados en la HAP refractaria o siempre que no se disponga de tratamientos médicos. Estos procedimientos sólo se deben realizar en centros especializados.
ENFERMEDADES ESPECÍFICAS
Hipertensión arterial pulmonar pediátrica
Incidencia. La prevalencia de la enfermedad cardíaca congénita es más elevada entre niños que entre adultos y es preciso tomar las oportunas precauciones para reconocer esta importante causa de HAP. Por el contrario, la prevalencia de la HAP asociada con ETC, hipertensión portal, infección por VIH y fármacos/toxinas es menor entre los niños. Aunque la HP persistente del recién nacido (HPPRN) está clasificada dentro de la HAP, su historia natural y su tratamiento son muy diferentes de las otras formas de HAP, por lo que se justifica su exclusión de este apartado. La HPPRN normalmente es transitoria171,172, el recién nacido se recupera completamente sin necesitad de terapia médica crónica o fallece durante el período neonatal aunque reciba los máximos cuidados terapéuticos cardiovasculares173.
Patogenia. No se han identificado diferencias claras entre los mecanismos que intervienen en el desarrollo de la HAP pediátrica o en adultos. Sin embargo, la HPPRN conlleva algunos mecanismos fisiopatológicos específicos causados por las características de la vasculatura pulmonar fetal y del foramen oval permeable174.
Además, la prevalencia de la vasorreactividad aguda es mayor en los niños con HAPI, lo que sugiere que la vasoconstricción puede prevalecer sobre los cambios vasculares obstructivos fijos en este subgrupo79,118.
Situación clínica y estudios diagnósticos. Los datos clínicos, diagnósticos y pronósticos sobre la HAP proceden fundamentalmente de la población adulta, por lo que a menudo es necesario extrapolar esta información para la población pediátrica.
Según el registro NIH, se ha observado que la mortalidad infantil es superior que la mortalidad en adultos en caso de ausencia de tratamiento. Sin embargo, los datos de este registro fueron recogidos de un pequeño número de casos pediátricos anteriores a las nuevas terapias médicas. Teóricamente, cabe esperar que la respuesta al tratamiento sea mejor en los niños, ya que su sistema vascular se remodela con el crecimiento. De hecho, los nuevos tratamientos médicos parecen tener claramente mejores resultados en niños que en adultos con HAP; sin embargo, el curso de la enfermedad es menos predecible175. No se han establecido las causas por las que un niño evoluciona de forma diferente a otro afectado por el mismo grado de HAP.
A pesar de que sólo disponemos de informes monocéntricos sobre la estrategia de diagnóstico, los niños con HP severa reciben los mismos estudios diagnósticos que se han descrito para adultos (véase el apartado Estrategia de diagnóstico)176. Estas pruebas incluyen la gasometría arterial, la medición de la saturación de oxígeno, el ecocardiograma, la gammagrafía de ventilación-perfusión, la TC torácica, la ecografía abdominal, los estudios sexológicos sobre la ETC, los estudios de hipercoagulabilidad y las pruebas de VIH. El diagnóstico debe ser confirmado mediante CCD.
Al igual que en los adultos con HAP severa, las pruebas de vasorreactividad pulmonar durante el CCD incluyen la evaluación de la respuesta aguda a un vasodilatador de acción rápida como el NO, el epoprostenol intravenoso o la adenosina intravenosa, para determinar la conveniencia de la terapia crónica con un BCC oral. La prevalencia de la vasorreactividad aguda es mayor en niños que en adultos y por ello se puede tratar de forma efectiva a más niños que adultos con BCC79,118.
Tratamiento. El algoritmo terapéutico para los niños con HAP es similar al utilizado en adultos; sin embargo, es preciso aclarar algunos temas específicos. Los niños que responden a una estrategia específica de tratamiento generalmente lo hacen mucho mejor que los adultos. Sin embargo, los niños que no responden a una terapia determinada presentan una supervivencia mucho menor que los adultos con enfermedad severa.
Debido a que el lecho vascular pulmonar del niño es más reactivo que el de los adultos, cualquier infección de las vías respiratorias que resulte en un desequilibrio de la ventilación-perfusión por hipoxia alveolar puede desencadenar eventos catastróficos si no se actúa de manera agresiva. Se recomienda la hospitalización de los niños con neumonía para la instauración de una terapia antibiótica, con administración de antitérmicos que eviten subidas de temperatura > 38 ºC (101 ºF) y minimizar las consecuencias del aumento de la demanda metabólica.
No se ha determinado si la anticoagulación indefinida es eficaz en los niños con HAP, ni tampoco si es segura y con un bajo perfil de riesgo/beneficio. Sin embargo, en la actualidad los expertos deciden anticoagular a los niños con insuficiencia cardíaca derecha.
La seguridad y la eficacia de los BCC está basada en la respuesta del paciente a la prueba de vasodilatación aguda; en el niño, la eficacia de este tratamiento es similar a la del adulto. Al igual que en el adulto, el régimen óptimo contempla dosis altas de BCC (véase el apartado Bloqueadores de los canales del calcio), pero el niño presenta mayor tolerancia y necesita una dosis más elevada por kilogramo.
Las indicaciones clínicas para la terapia indefinida con epoprostenol intravenoso es similar en niños y adultos, pero no se ha establecido la dosis óptima en ninguno de los 2 grupos. La dosis de inicio del tratamiento es de 2 ng/kg/min para ambos grupos y su incremento depende de las necesidades, aunque el aumento suele ser rápido durante los primeros meses tras la instauración del tratamiento. Para el paciente adulto con HAP, la dosis media al año es de aproximadamente de 20-40 ng/kg/min, mientras que la dosis pediátrica al año, sobre todo en niños pequeños, se acerca a los 50-80 ng/kg/min; sin embargo, la dosis «óptima» varía de manera considerable entre pacientes.
Se ha utilizado el beraprost oral, el iloprost inhalado y la infusión subcutánea de treprostinil en el tratamiento de niños con HAP, alcanzando distintos grados de éxito. En la práctica puede resultar difícil la administración de la dosis efectiva de iloprost inhalado (aún cuando el niño coopere) y la infusión subcutánea de treprostinil puede ser demasiado dolorosa. La experiencia sugiere que, al igual que sucede con el epoprostenol intravenoso, los niños necesitan una dosis más alta por kilogramo que los adultos.
Se ha realizado un estudio no controlado, abierto y con dosis única o múltiple en niños de 4-17 años de edad con HAP (BREATHE-3) para valorar las propiedades farmacocinéticas, la tolerancia y la seguridad del bosentán oral. En este estudio preliminar se observó una mejoría significativa de los parámetros hemodinámicos en los 18 niños participantes tras 12 semanas de tratamiento con bosentán solo o combinado con epoprostenol177.
Se ha descrito el uso de inhibidores de la fosfodiesterasa-5, como el sildenafilo, pero los datos disponibles se limitan a series de casos demasiado reducidas178.
Hipertensión arterial pulmonar asociada con el síndrome de Eisenmenger
Incidencia. Consultar el síndrome de Eisenmenger en la clasificación de los cortocircuitos sistémico-pulmonares congénitos.
Patogenia. El síndrome de Eisenmenger se define como el defecto cardíaco congénito que causa inicialmente un cortocircuito importante izquierda-derecha que induce enfermedad vascular pulmonar severa e HAP y que resulta en la inversión del sentido del cortocircuito179. Con el cortocircuito izquierda-derecha inicial, la exposición de la vasculatura pulmonar al incremento del flujo sanguíneo y de la presión puede dar como resultado la presentación de la enfermedad vascular obstructiva y, en momento en que la RVP se acerca o sobrepasa la resistencia sistémica, el cortocircuito se invierte.
Situación clínica y estudios diagnósticos. La mayoría de los pacientes presentan afección de la tolerancia al ejercicio y disnea de esfuerzo, pero estos síntomas pueden estar bien compensados durante años. Puede aparecer hemoptisis como consecuencia de la rotura de arterias bronquiales dilatadas. Debido a que los pacientes con una saturación arterial de oxígeno reducida presentan hemostasis anormal, tienen riesgo de sangrado y trombosis. Los accidentes cerebrovasculares pueden ocurrir como resultado de la embolización paradójica, de la trombosis venosa de los vasos cerebrales y de hemorragias intracraneales. Además, los pacientes con esta enfermedad tienen riesgo de abscesos cerebrales. Los pacientes con el síndrome de Eisenmenger pueden tener síncopes debidos a un gasto cardíaco inadecuado o, con menos frecuencia, una arritmia. Los síntomas de insuficiencia cardíaca, poco comunes hasta que la enfermedad está en un estado avanzado, conllevan un mal pronóstico. La supervivencia de los pacientes con el síndrome de Eisenmenger es mejor que la de los sujetos con HAPI o HAPA, en una clase funcional comparable. En una serie de 100 pacientes en espera de trasplante, la supervivencia actuarial de los pacientes que no recibieron trasplante fue del 97% al año, del 89% a los 2 años y del 77% a los 3 años en los pacientes con síndrome de Eisenmenger, y del 77, el 69 y el 35%, respectivamente, en los pacientes con HAPI180.
Tratamiento. En los pacientes con el síndrome de Eisenmenger, la recomendación de tratamiento está basada fundamentalmente en la experiencia de los expertos y no en ensayos clínicos específicos106,181. La flebotomía con reemplazamiento isovolumétrico se debe realizar en pacientes con síntomas moderados o severos de hiperviscosidad (cefaleas y falta de concentración) que normalmente están presentes cuando el hematocrito es > 65%; no se debe realizar en pacientes asintomáticos o con sintomatología leve (con independencia de los valores del hematocrito). Normalmente, se alivian los síntomas con la retirada de una unidad de sangre, que siempre se reemplaza con el mismo volumen de dextrosa o salino106. No se deben realizar flebotomías más de 2-3 veces al año para evitar el agotamiento de las reservas de hierro y la producción de células hipocrómicas que aumenten la viscosidad de la sangre. En caso de signos de insuficiencia cardíaca derecha se pueden administrar diuréticos.
El uso de la terapia suplementaria de oxígeno es controvertida182 y sólo se utilizará en casos en los que pueda producir un aumento importante de la saturación arterial y/o una mejora del estado clínico del paciente (componente pulmonar restrictivo). En algunos centros, los pacientes con el síndrome de Eisenmenger, al igual que otros sujetos con HAP, reciben terapia anticoagulante siempre que no haya contraindicaciones. Otros autores prefieren evitar este tratamiento, ya que podría exacerbar la diátesis hemorrágica183.
Lamentablemente, pocos ensayos clínicos sobre el efecto de los nuevos tratamientos médicos para la HAP han incluido a pacientes con el síndrome de Eisenmenger127,130,133. Una de las razones para ello es que la evolución natural del síndrome de Eisenmenger no tratado, a pesar de ser significativamente peor que el de la población normal, en la mayoría de los casos tiene una progresión muy lenta, lo que dificulta la realización de ensayos clínicos. Aunque la evolución natural de la HAPI y de la HAP asociada con enfermedades cardíacas congénitas es muy diferente, la similitud de su histopatología permite pensar que una estrategia común de tratamiento podría ser adecuada y efectiva. Sin embargo, es necesario probar científicamente la eficacia de los nuevos tratamientos para determinar la relación riesgo/beneficio. Se ha demostrado que el uso de epoprostenol intravenoso produce efectos favorables en la hemodinámica y en la capacidad de ejercicio125, y los efectos del treprostinil subcutáneo en pacientes con el síndrome de Eisenmenger no son diferentes de los observados en los pacientes con HAPI132. Actualmente se está desarrollando un ensayo clínico aleatorizado (BREATHE-5) para valorar los efectos del bosentán en 65 pacientes con el síndrome de Eisenmenger.
El trasplante de pulmón con reparación del defecto cardíaco o el trasplante corazón-pulmón combinado es una opción de tratamiento para los pacientes con el síndrome de Eisenmenger que presentan marcadores de mal pronóstico (síncope, insuficiencia cardíaca derecha refractaria, clase funcional III o IV de la NYHA o hipoxemia severa). Debido a que el éxito del trasplante de pulmón es relativamente limitado y a la razonablemente buena supervivencia de los pacientes con tratamiento médico, es imperativo extremar la precaución en la selección de pacientes para el trasplante.
Hipertensión portopulmonar
Incidencia. La HAP es una complicación ampliamente conocida de las enfermedades hepáticas crónicas184-186. La hipertensión portal, más que una enfermedad hepática en sí misma parece más bien el mayor factor de riesgo determinante de la evolución de la HP, de ahí el concepto de hipertensión portopulmonar185. La hipertensión portopulmonar fue descrita inicialmente por Mantz y Craige en 1951; sin embargo y debido a su baja incidencia, tuvieron que pasar muchos años antes de que se pudiese determinar si esta asociación era fortuita o causal. Hay evidencias que sugieren que la presencia de hipertensión portal y el desarrollo de HAP no es una simple coincidencia185,187. Y, en efecto, la incidencia de la HAP en pacientes con hipertensión portal es mucho más elevada que la incidencia estimada de la HAPI en la población general. En un estudio restrospectivo post mortem se observó que la incidencia de HAP en la totalidad de los casos en los que se realizó la autopsia fue del 0,13%, frente al 0,73% en los casos en los que los pacientes habían padecido cirrosis e hipertensión portal.
En 2 estudios hemodinámicos prospectivos se observó que el 2% de los pacientes con cirrosis e hipertensión portal presentó HAP significativa. En otros 2 estudios realizados en pacientes que recibieron trasplante hepático se demostró una prevalencia de la hipertensión pulmonar del 4 y del 3,5%, respectivamente.
Por último, en el estudio IPPHS se confirmó que la cirrosis era un factor de riesgo para la HAP188. La hipertensión portal no es una causa rara de HAP; en el registro NIH, el porcentaje de pacientes con hipertensión portopulmonar fue del 8%. Los cortocircuitos portosistémicos quirúrgicos aumentan la incidencia de HP en los pacientes con hipertensión portal, como se demostró en un estudio retrospectivo, en el que aproximadamente al 65% de los pacientes con HAP se le habían realizado cortocircuitos quirúrgicos, mientras que al 35% no189. Estos hallazgos sugieren claramente que el desarrollo de la HAP en pacientes con hipertensión portal está relacionado con el desarrollo de cortocircuitos portosistémicos, más que con la hipertensión portal por sí misma. La presencia de enfermedad hepática parenquimatosa y su severidad no están asociadas con el riesgo de HAP debido a que esta complicación ocurre en pacientes con hipertensión portal extrahepática189. De igual forma, el grado de hipertensión portal, estimada por el gradiente de presión hepático-venosa y por los cambios hemodinámicos, no está asociado con el desarrollo de la HAP184. Solamente la duración de la hipertensión portal puede aumentar el riesgo de desarrollo de HAP.
Patogenia. Todavía se desconoce el mecanismo por el cual la hipertensión portal favorece el desarrollo de la HAP185. La presencia de un cortocircuito portosistémico puede favorecer que sustancias vasoconstrictoras y vasoproliferativas, normalmente aclaradas por el hígado, lleguen a la circulación pulmonar. La serotonina producida por las células enterocromafines intestinales puede ser una de estas sustancias. Los hallazgos histopatológicos de la hipertensión portopulmonar no se diferencian de los encontrados normalmente en la HAPI190.
Situación clínica y estudios diagnósticos. El estado clínico de los pacientes con hipertensión portopulmonar posiblemente no se diferencie del estado clínico de los pacientes con HAPI o puede incluir algunos signos y síntomas de la enfermedad hepática subyacente185.
Es conveniente realizar estudios ecocardiográficos sistemáticos para la detección de la HP en los pacientes sintomáticos con enfermedades hepáticas y/o candidatos a trasplante de hígado. En todos los casos en los que se observe un incremento de la PAPS se realizará un CCD que permita determinar los cambios hemodinámicos subyacentes, así como definir el pronóstico y sus implicaciones terapéuticas.
En cuanto a los parámetros hemodinámicos, los pacientes con hipertensión portopulmonar, frente a pacientes con HAPI, presentan un gasto cardíaco mayor y una resistencia vascular sistémica y pulmonar significativamente menor191. El diagnóstico de la hipertensión portal mediante un catéter Swan-Ganz durante el CCD requiere la determinación de un gradiente de presión entre las venas hepáticas libres y ocluidas (enclavamiento) o un gradiente de presión hepático-venosa > 10 mmHg (valor normal < 5 mmHg)67.
En un estudio retrospectivo185, los pacientes con hipertensión portopulmonar mostraron una tasa mayor de supervivencia que los pacientes con HAPI, aunque hay cierta polémica sobre este tema192.
Tratamiento. El tratamiento de la hipertensión portopulmonar presenta ciertos retos y no ha sido completamente estudiado. Se puede utilizar oxígeno complementario, si fuera necesario, para mantener unas saturaciones arteriales de oxígeno > 90%. La terapia con diuréticos puede ser utilizada para controlar la sobrecarga del volumen, los edemas y la ascitis. En esta población, la terapia anticoagulante no ha sido estudiada en profundidad y, posiblemente, debe ser evitada en los pacientes con la función hepática afectada y recuento plaquetario bajo, así como en los pacientes con mayor riesgo de sangrado debido a la presencia de varices gastroesofágicas. En ausencia de un marcado aumento del gasto cardíaco y con una presión vascular pulmonar relativamente baja, los pacientes con HP leve o moderada deben ser sometidos al test vasodilatador agudo en el laboratorio de hemodinámica. En caso de respuesta aguda positiva al agente vasodilatador, se debe considerar la instauración cuidadosa de un BCC. Los bloqueadores beta que se utilizan normalmente en el tratamiento de la hipertensión portal y en la reducción del riesgo de sangrado por varices suelen ser mal tolerados en los casos de HAP asociada debido al efecto inotrópico negativo sobre el miocardio ventricular derecho.
Se ha descrito el tratamiento con epoprostenol intravenoso para la hipertensión portopulmonar en comunicaciones de series reducidas o casos aislados188,193,194. Parece que los pacientes con hipertensión portopulmonar responden al tratamiento crónico con epoprostenol intravenoso de forma similar a los pacientes con HAPI; sin embargo, se ha comunicado una mayor incidencia de la ascitis y esplenomegalia195.
La HAP significativa puede aumentar de manera considerable el riesgo asociado al trasplante hepático y, generalmente, la HAP constituye una contraindicación al trasplante cuando la PAP media es ≥ 35 mmHg y/o la RVP es ≥ 250 dinas/s/cm196. En algunos casos se pueden reducir la PAP media y la RVP mediante un tratamiento agresivo, que incluye el epoprostenol, para conseguir que el paciente con HAP pase de ser un candidato limítrofe a ser un candidato aceptable para el trasplante hepático192. En los casos en los que la severidad de la enfermedad requiera un trasplante multiórgano, como el trasplante de hígado y pulmón o corazón-pulmón, se considera que el riesgo es muy elevado198.
En algunos pacientes se ha observado una mejoría de la HAP tras el trasplante hepático199, especialmente en los que presentan un gasto cardíaco elevado antes del trasplante, que disminuye tras el éxito de éste. Otros pacientes pueden experimentar un empeoramiento de la HAP después del trasplante hepático. En ocasiones se puede suspender el tratamiento con epoprostenol intravenoso en el paciente trasplantado, pero es preciso hacerlo muy gradualmente y bajo estrecha vigilancia médica.
Debido a su hepatotoxicidad, la mayoría de los expertos recomendarían posiblemente evitar los antagonistas orales de la endotelina, como el bosentán, en esta población de pacientes. A pesar de los buenos resultados observados en algunas series de casos realizados en centros especializados, la relación riesgo/beneficio del tratamiento con antagonistas de los receptores de la endotelina en pacientes con enfermedad hepática debe ser evaluado a largo plazo.
Hipertensión arterial pulmonar asociada con la infección por VIH
Incidencia. La HAP es una complicación rara pero bien documentada de la infección por VIH; se han comunicado más de 200 casos en la bibliografía19,200,201. Actualmente, las manifestaciones cardiovasculares no infecciosas de la infección por VIH, tales como la miocardiopatía dilatada, el derrame pericárdico, la endocarditis trombótica no bacteriana, la aterosclerosis acelerada y la HAP, se detectan con más frecuencia como consecuencia de una mayor supervivencia y de una profilaxis más adecuada frente a las infecciones oportunistas202. En un importante estudio de casos y controles, 3.349 pacientes infectados por el VIH durante un período de 5,5 años mostraron una incidencia de la HP de 0,57%, con el resultado de una incidencia anual del 0,1%203.
Patogenia. Se desconoce el mecanismo de desarrollo de la HAP, aunque se sospecha una acción indirecta del VIH a través de segundos mensajeros, como las citocinas204, los factores de crecimiento204 o la ET-1205, sobre la base de la ausencia de ADN viral en las células endoteliales pulmonares204,206. Esta hipótesis está reforzada por la presencia de células inflamatorias perivasculares en la HAP asociada con el VIH207,208. Además, se cree también que la predisposición genética desempeña un papel importante, ya que esta complicación afecta solamente a una minoría de los pacientes infectados por el VIH. La ausencia de mutaciones de la BMPR2 en un subgrupo de pacientes209 con HAP asociada con el VIH sugiere que están implicados otros factores de susceptibilidad.
Situación clínica y estudios diagnósticos. La HAP asociada con la infección por VIH presenta los mismos hallazgos clínicos, hemodinámicos e histopatológicos de la HAPI y no parece estar relacionada con la vía de transmisión del VIH ni con el grado de inmunodepresión210. Los pacientes con VIH con frecuencia también están infectados por los virus de la hepatitis B y C y, por tanto, la presencia de enfermedad hepática concomitante es probable.
En los pacientes sintomáticos infectados por el VIH es preciso realizar estudios ecocardiográficos para la detección de la HP. Además, se hace necesario descartar otras causas de HP, como la enfermedad cardíaca izquierda, la enfermedad parenquimatosa pulmonar y la enfermedad hepática.
Se recomienda la realización de un cateterismo cardíaco derecho en tod en los que haya sospecha de HAP asociada con la infección por VIH, con el objeto de confirmar el diagnóstico, determinar la severidad y descartar enfermedad cardíaca izquierda.
La mortalidad de los pacientes con HAP asociada con el VIH está relacionada fundamentalmente con la HAP en sí misma, más con otras complicaciones de la infección por VIH210; la HAP es un factor independiente de predicción de la mortalidad en este grupo de pacientes203.
Tratamiento. En la HAP asociada con el VIH, las opciones terapéuticas están menos establecidas que en otras formas de HAP. Con frecuencia, la anticoagulación oral está contraindicada debido a un recuento plaquetario reducido, a la dificultad de administración del tratamiento y a la posible interacción entre las medicación del VIH y la warfarina (cumarínicos).
En relación con el test vasodilatador agudo y el efecto beneficioso del tratamiento con BCC, no se dispone de información publicada sobre este grupo de pacientes. En un estudio abierto y no controlado realizado en 6 pacientes con HAP asociada con el VIH128 se observó que la infusión continua de epoprostenol puede ser efectiva para mejorar el estado funcional y los parámetros hemodinámicos hasta 12-47 meses. En este grupo de población no se recomienda el trasplante de pulmón.
Todavía está por establecer el papel de una terapia antirretroviral altamente activa en el manejo de la HAP asociada con el VIH. Se ha observado un efecto beneficioso en la hemodinámica pulmonar en los pacientes tratados con inhibidores de la transcriptasa inversa de nucleósidos203. Recientemente se ha comunicado 1 caso en el que se observó una mejoría hemodinámica mantenida a largo plazo con este tratamiento, sin el uso asociado de agentes vasodilatadores211. Por último, en una serie de casos de un solo centro en la que se incluía a 82 pacientes209, el análisis univariable indicó que el recuento de linfocitos CD4 (> 212 células/µl), la combinación de la terapia antirretroviral y el uso del epoprostenol (infusión) estaban asociados con una mejoría de la supervivencia. En el análisis multivariable, sólo el recuento de linfocitos CD4 constituía un factor de predicción independiente de la mortalidad, seguramente porque, en la población del estudio, la terapia antirretroviral combinada y la infusión de epoprostenol estaban muy relacionadas.
De manera más reciente, en una serie de 16 pacientes con HAP asociada con el VIH se observaron resultados clínicos y hemodinámicos favorables con el tratamiento con bosentán212.
En resumen, los estudios no controlados sugieren que los pacientes con HAP severa asociada con el VIH pueden responder de manera favorable a la terapia antirretroviral combinada, al epoprostenol y, posiblemente, al bosentán. Sin embargo, el uso de epoprostenol, antagonistas de los receptores de la endotelina e inhibidores de la fosfodiesterasa tipo 5 (FDD5) todavía debe ser evaluado mediante ensayos clínicos controlados en este grupo de pacientes213.
Hipertensión arterial pulmonar asociada con enfermedades del tejido conectivo
Incidencia. La HP es una complicación bien conocida de las enfermedades del tejido conectivo (ETC), tales como la esclerosis sistémica214, el lupus eritematoso sistémico215, la ETC mixta216 y, en menor grado, la artritis reumatoide, la dermatopolimiositis y el síndrome primario de Sjögren217. En estos pacientes, la HAP puede presentarse asociada con la fibrosis intersticial o como consecuencia de la proliferación vascular directa en ausencia de enfermedad parenquimatosa significativa o de hipoxia crónica. Además, también puede estar presente la hipertensión pulmonar venosa subsiguiente a la enfermedad cardíaca izquierda. Es de suma importancia determinar cuál es el factor operativo, ya que el tratamiento puede ser muy distinto dependiendo de cada proceso.
Resulta difícil calcular la prevalencia de la HAP en los pacientes con ETC debido a la escasez de datos epidemiológicos. La prevalencia estimada de la HAP en estos pacientes es muy variable y depende de la definición de la HAP, del método utilizado para evaluar la PAP y del sesgo potencial de la población del estudio4.
La esclerosis sistémica, sobre todo en su variante limitada, definida anteriormente como síndrome de CREST (calcinosis, enfermedad de Raynaud, alteraciones esofágicas, esclerodactilia y telangiectasia), representa la mayor ETC asociada con la HAP. En un registro de 722 pacientes que se ha completado recientemente en el Reino Unido sobre la HP en pacientes con esclerosis sistémica, se observó una prevalencia de alrededor del 12%214. En otras series de 930 pacientes con esclerosis sistémica, la incidencia acumulativa de la HP fue del 13%49. Sin embargo, en otro estudio enfocado según la población, la prevalencia de la HP fue del 2,6% en 3.778 pacientes218. En el registro NIH, en 236 casos de HAP no explicada, 18 casos estaban asociados con la ETC48. En varios centros especializados en la HAP, más del 10% de los pacientes con HAP severa presentaba ETC asociada, en la mayor parte de los casos se trataba de la variante CREST de la esclerodermia.
Patogenia. Los cambios histopatológicos de la HAP asociada con la ETC generalmente no se diferencian de los cambios propios de la HAPI. Es más, en estos pacientes se ha descrito el espectro total de la patología vascular pulmonar, incluida la EVOP y la HCP. Los mecanismos fisiopatológicos que desencadenan la HAP en los pacientes con enfermedades del tejido conectivo se desconocen. El llamado fenómeno de Raynaud (por vasospasmo pulmonar) podría, hipotéticamente, estar implicado. La presencia de anticuerpos antinucleares, factor reumatoide, inmunoglobulina G y depósitos de fracciones del complemento en la pared de los vasos pulmonares sugieren la intervención de mecanismos inmunológicos.
Situación clínica y estudios diagnósticos. En comparación con los pacientes con HAPI, los pacientes con HAP asociada con ETC son mayores, sobre todo mujeres, presentan un gasto cardíaco significativamente más bajo y muestran una tendencia hacia una supervivencia más corta. En un registro del Reino Unido se comunicó un período medio entre el diagnóstico de la esclerosis sistémica y la HAP de 14 años; asimismo, la enfermedad se encontró, por lo general, en pacientes de edad media avanzada (media de edad de 66 años).
Los síntomas y la presentación clínica son muy similares a los de la HAPI y, en algunas ocasiones, se puede identificar a los pacientes con ETC asociada mediante pruebas inmunológicas sistemáticas. La TC de alta resolución es útil como prueba de exclusión para la presencia/ausencia de fibrosis significativa. Se ha confirmado que la mortalidad es más elevada que en la HAPI (un 40% de mortalidad al año para los pacientes con enfermedad en estado avanzado) y los predictores de resultados fueron los mismos que en la HAPI (PAD, PAP e índice cardíaco).
Se ha sugerido la conveniencia de realizar estudios ecocardiográficos anuales para la detección de la HP en pacientes asintomáticos con enfermedades del espectro de la esclerodermia49 y sólo en presencia de síntomas en otras enfermedades del tejido conectivo. Sin embargo, no están claras las razones que justifiquen el estudio ecocardiográfico preventivo en los pacientes asintomáticos, ya que no tenemos evidencia de la efectividad de los tratamientos en este subgrupo. En cualquier caso, la detección temprana de síntomas relacionados con la HAP requiere una valoración ecocardiográfica, inmediata y meticulosa, de cualquier paciente con ETC y en cualquier momento.
Al igual que en otras formas de la HAP, se recomienda la realización de CCD en todos los casos en los que haya sospecha de HAP asociada con ETC para confirmar el diagnóstico, determinar el grado de severidad y descartar enfermedad de cavidades izquierdas.
Tratamiento. El tratamiento de los pacientes con HAP asociada con la ETC es más complejo que el tratamiento de la HAPI. La terapia inmunodepresora sólo es efectiva en una minoría de pacientes que, fundamentalmente, presentan enfermedades no pertenecientes al espectro de la esclerodermia.
La tasa de respuesta a la prueba al test vasodilatador agudo y de respuesta favorable al tratamiento con BCC es más baja que en la HAPI. Además, no se ha establecido con claridad la relación riesgo/beneficio de la anticoagulación oral.
En un estudio aleatorizado y con una duración de 3 meses se demostró que la terapia continua con epoprostenol mejora la capacidad de ejercicio, los síntomas y la hemodinámica de los pacientes con enfermedades del espectro de la esclerodermia113. En este estudio no se documentó una mejoría de la supervivencia. Algunos análisis retrospectivos muestran que el efecto del epoprostenol intravenoso en la supervivencia de los pacientes con HAPI es mejor que en los pacientes con esclerodermia218,220.
En un estudio aleatorizado sobre la HAP se evaluó la administración subcutánea continua de treprostinil en un subgrupo de 90 pacientes con HAP y ETC, incluidos el lupus eritematoso sistémico, la esclerodermia difusa, la esclerodermia limitada y la ETC mixta/síndrome de superposición. Después de 12 semanas de tratamiento se observó una mejoría en la capacidad de ejercicio, en los síntomas de la HAP y en la hemodinámica. Entre los eventos adversos se contaron el dolor en el punto de punción y los efectos secundarios típicos relacionados con las prostaglandinas221.
En un estudio aleatorizado, doble ciego, de 12 semanas, en el que se incluía un subgrupo de 47 pacientes con ETC, se observó que el bosentán mejoró significativamente la capacidad de ejercicio frente al placebo en esta población. Sin embargo, aunque se obtuvo un efecto similar en los pacientes con HAPI y con HAP asociada con esclerodermia, el bosentán mejoró la distancia caminada en los pacientes con HAPI (+46 m en el grupo con bosentán frente a -5 m en el grupo placebo), mientras que en el grupo de pacientes con esclerodermia sólo evitó la reducción de la distancia caminada (+3 m en el grupo con bosentán frente a -40 m en el grupo placebo)144.
En resumen, la respuesta a los tratamientos y la supervivencia a largo plazo parecen peores en los pacientes con HAP asociada con la ETC que en los pacientes con HAPI.
Enfermedad venooclusiva pulmonar y hemangiomatosis capilar pulmonar
Incidencia. Tanto la enfermedad venooclusiva pulmonar (EVOP) como la hemangiomatosis capilar pulmonar (HCP) son enfermedades poco comunes que, sin embargo, de forma creciente se reconocen como causas de la HAP222. En la bibliografía se han comunicado menos de 200 casos de EVOP y HCP.
Patogenia. Como se ha discutido en la clasificación clínica y en la patología (ver secciones), la EVOP y la HCP son similares en algunos aspectos, especialmente en los relacionados con los cambios en el parénquima pulmonar (hemosiderosis, edema intersticial y dilatación linfática), con la fibrosis de la íntima arterial pulmonar y con la hipertrofia de la media6.
Los informes sobre la incidencia familiar, tanto de la EVOP y la HCP223 como de la HAP, tienen especial interés. Por último, la mutación del BMPR2, el gen asociado con la HAP familiar y con la HAPI, ha sido documentada en 1 paciente con EVOP224. Estos hallazgos sugieren que la EVOP, la HCP y la HAP pueden formar parte del espectro de una misma enfermedad.
Situación clínica y estudios diagnósticos. Con frecuencia, la presentación clínica de estos pacientes no se diferencia de la de los pacientes con HAPI. Sin embargo, en la exploración física se pueden encontrar signos sugestivos de un diagnóstico diferente de la HAPI, como dedos en palillo de tambor (acropaquia) y/o crepitantes basales en la auscultación torácica. Las series de casos indican que la EVOP/HCP está asociada con una hipoxemia más severa y con una reducción de la capacidad de difusión del monóxido de carbono (DLCO) en una inspiración, mientras que la espirometría y el volumen pulmonar se mantienen dentro de límites normales. La reducción significativa en la DLCO, que se observa con frecuencia, puede ser explicada por el edema intersticial crónico secundario a la obstrucción venosa pulmonar.
Los datos hemodinámicos de la EVOP/HCP y la HAPI son similares, aunque en algunos pacientes la hipoxemia es desproporcionada en relación con el grado de HAP y la disfunción cardíaca derecha. Curiosamente, la PCP suele ser normal a pesar de la participación poscapilar; de hecho, los cambios patológicos ocurren normalmente en las vénulas, a menudo sin participación de las venas más grandes. La columna estática de sangre que se produce durante la medición de la PCP no se ve alterada por los cambios en las venas pulmonares pequeñas, siempre que se mantenga la conexión con las venas más grandes e inalteradas, que es donde se mide la presión en el segmento arterial ocluido. Los datos radiológicos son de gran ayuda para la detección de la EVOP/HCP62,225-227. La presencia de líneas B de Kerley, derrame pleural e irregularidades poco uniformes observadas en la radiografía de tórax estándar pueden proporcionar pistas importantes para el diagnóstico. La TC torácica (de cortes finos) revela cambios característicos. Los hallazgos más frecuentes son los patrones centrilobulares no uniformes, las líneas engrosadas del septo, el derrame pleural y las adenopatías mediastínicas. Estas anomalías tienen una correlación importante con el desarrollo del edema pulmonar tras la infusión de epoprostenol intravenoso: las opacidades en forma de reloj de arena fueron significativamente más frecuentes en la EVOP/HCP que en la HAPI (p = 0,003). En la EVOP/HCP, estas opacidades fueron abundantes y presentaban una distribución aleatoria. Las características morfológicas de la opacidad son importantes. La distribución centrilobular (opacidad nodular centrilobular difusa) es más frecuente en la EVOP/HCP (p = 0,03). Por el contrario, la distribución panlobular (regiones geográficas de atenuación pulmonar con bordes relativamente bien definidos) fue observada en ambos grupos y no es predictiva. Las líneas septales subpleurales (p < 0,0001) y la adenopatía (p < 0,0001) son también significativamente más frecuentes en la EVOP/HCP que en la HAPI. La asociación de estos tres hallazgos fue específica de la EVOP en casos de HP (especificidad del 100%) con un 66% de sensibilidad. Por lo tanto, la presencia y asociación de opacidades en forma de reloj de arena (especialmente con distribución centrilobular), líneas septales y adenopatía en la TC de tórax son indicativas de EVOP/HCP en pacientes con HAP aparente. En presencia de dichas anormalidades radiológicas se tomarán precauciones antes de la instauración de una terapia vasodilatadora.
Entre otras pruebas para el diagnóstico de la EVOP se incluye la broncoscopia con lavado broncoalveolar. Comparada con la HAPI, la EVOP/HCP se caracteriza por una elevación significativa del recuento celular del lavado broncoalveolar. Sin embargo, el porcentaje de macrófagos, linfocitos y neutrófilos es similar. Debido a que la EVOP/HCP afecta a la vasculatura poscapilar, generalmente se asocia con una hemorragia alveolar oculta y con macrófagos cargados de hemosiderina. En series recientes de casos, el porcentaje de macrófagos cargados de hemosiderina fue mayor en la EVOP que en la HAPI (54 ± 37% frente a 3 ± 6%; p = 0,0006). La escala de Golde fue notablemente más elevada en la EVOP (109 ± 97 frente a 4 ± 10; p = 0,0004)228. Para concluir, la evidencia combinada de HAP, números elevados de macrófagos alveolares cargados de hemosiderina e infiltrados del intersticio pulmonar sugieren el diagnóstico de EVOP/HCP.
Tratamiento. En la nueva clasificación clínica, la EVOP y la HCP están incluidas en la categoría de la HAP asociada con una afección pulmonar venosa o capilar significativa. Este subgrupo probablemente requiere un manejo similar a otros subgrupos de la HAP. Sin embargo, el pronóstico es peor y el curso de la enfermedad suele ser más rápido. Además, es necesario tener mucha precaución con el uso de vasodilatadores, especialmente el epoprostenol, debido al alto riesgo de edema pulmonar229,230. Sin embargo, se ha comunicado una mejoría clínica sustancial en pacientes individuales tratados con esta medicación. No se dispone de datos relativos al uso de nuevas terapias (como los antagonistas de los receptores de la endotelina) en el tratamiento de la EVOP/HCP. El tratamiento médico de estos pacientes sólo debe ser realizado en centros con amplia experiencia en el diagnóstico y manejo de la HAP. Los pacientes deben ser informados de los riesgos antes de la instauración del tratamiento. Se puede considerar la septostomía auricular aunque se encuentra limitada por la hipoxemia, más común en la EVOP/HCP que en otras clases de HAP. La única terapia curativa para la EVOP/HCP es el trasplante de pulmón y, al igual que sucede en la HAPI, no se ha comunicado la recurrencia de la enfermedad tras el trasplante.
AGRADECIMIENTOS
El CPG quiere expresar su agradecimiento a los revisores de la primera versión de esta guía titulada «Guía de Práctica Clínica de la ESC de la hipertensión pulmonar primaria»: Joern Carlsen (Dinamarca), Sean Gaine (Irlanda), Stefano Ghio (Italia), Marc Humbert (Francia), Irene Lang (Austria), Patrizia Presbitero (Italia) y Pietro Zonzin (Italia).
APÉNDICE A. LISTA DE ABREVIATURAS
BCC: bloqueadores de los canales de calcio.
BMPR2: receptor 2 de la proteína morfogenética ósea.
CCD: cateterismo cardíaco derecho.
DLCO: capacidad de difusión del monóxido de carbono.
ECA: ensayos clínicos aleatorizados y controlados.
EIP: enfermedad instersticial pulmonar.
EMEA: Agencia Europea para la Evaluación de Productos Médicos.
ET-1: endotelina 1.
ETC: enfermedad del tejido conectivo.
ETE: ecocardiografía transesofágica.
ETT: ecocardiografía transtorácica.
EVOP: enfermedad venooclusiva pulmonar.
FDA: Food and Drug Administration.
FDE: fosfodiesterasa.
GMPc: monofosfato de guanosina 3o-5o cíclico.
HAP: hipertensión arterial pulmonar.
HAPA: hipertensión arterial pulmonar relacionada con factores de riesgo o enfermedades asociadas.
HAPF: hipertensión arterial pulmonar familiar.
HAPI: hipertensión arterial pulmonar idiopática.
HCP: hemangiomatosis capilar pulmonar.
HP: hipertensión pulmonar.
HPP: hipertensión pulmonar primaria.
HPPRN: hipertensión pulmonar persistente del recién nacido.
HPTEC: hipertensión pulmonar por tromboembolia crónica.
I.V.: intravenoso.
NO: óxido nítrico.
NYHA: New York Heart Association.
OMS: Organización Mundial de la Salud.
PAD: presión de la aurícula derecha.
PAP: presión arterial pulmonar.
PAPS: presión arterial pulmonar sistólica.
PCP: p resión de enclavamiento pulmonar.
PECP: prueba de esfuerzo cardiopulmonar.
PFP: pruebas de la función pulmonar.
PSVD: presión sistólica del ventrículo derecho.
RVP: resistencia vascular pulmonar.
TC: tomografía computarizada.
TCAR: tomografía computarizada de alta resolución.
TxA2: tromboxano A2.
V/Q: ventilación/perfusión.
VIH: virus de inmunodeficiencia humana.
Los comentarios-anotaciones (*) incluidos en esta traducción de las Guías han sido realizados por la Dra. Pilar Escribano (Madrid, España).
(*) La indicación de referir al paciente con hipertensión pulmonar a un centro con experiencia en el manejo de esta enfermedad está también incluido (grado de recomendación alta) en las recientes guías del American College of Chest Physicians1, dada la complejidad de los procedimientos diagnósticos y terapéuticos.
1. Badesh D, Ahman S, Ahearn G, Barst G, Mc Grory D, Simonneau G, et al. Medical therapy for pulmonary arterial hypertension. ACCP Evidence-Based Clinical Guideliness. Chest. 2004;126:S35-62.
aCorrespondencia.
Coordinador: Nazzareno Galiè MD, Institute of Cardiology, University of Bologna, Via Massarenti, 9, 40138 Bologna, Italy. Tel.: + 39 051 349858; fax: +39 051 344859.
Correo electrónico: n.galie@bo.nettuno.it,
amanes@orsolamalpighi.med.unibo.it (N. Galiè).