Cuando se celebró en Ginebra la reunión de la Organización Mundial de la Salud (OMS) sobre «hipertensión pulmonar primaria» en 1973, esta entidad se consideraba una enfermedad huérfana y, al no haber tratamientos médicos eficaces, su diagnóstico constituía una sentencia de muerte para la mayoría de los pacientes1. Actualmente se reconoce de manera general que la hipertensión pulmonar (HP) es un trastorno frecuente, que afecta a alrededor del 1% de la población mundial y hasta un 10% de los mayores de 65 años2. Sin embargo, la mayor parte de los casos de HP se asocian a enfermedades crónicas pulmonares o del corazón izquierdo2–4, y la hipertensión arterial pulmonar (HAP), que corresponde al grupo 1 de la clasificación clínica actual4,5, continúa siendo una entidad bastante infrecuente. La prevalencia descrita de la HAP idiopática (HAPI), que equivale a la «hipertensión pulmonar primaria» de la época inicial, es actualmente de entre 15 y 26 casos por millón de habitantes4,6.
En las últimas 2 décadas, se han realizado enormes esfuerzos por mejorar el conocimiento de la fisiopatología de la HAP. Así, se han identificado las vías de la endotelina, el óxido nítrico y la prostaciclina como importantes factores contribuyentes a la aparición y la progresión de la enfermedad que son dianas relevantes para las intervenciones terapéuticas7. Como resultado de ello, se desarrollaron numerosos tratamientos dirigidos a la HAP que se han aplicado en monoterapia o en combinación8. Además del desarrollo y la autorización de nuevos fármacos para la HAP, se confirma una tendencia a llevar a cabo estudios de resultados clínicos a largo plazo mediante el diseño de ensayos clínicos que utilizan variables de valoración combinadas de morbilidad y mortalidad9–11. Algunos análisis recientes indican que las estrategias de tratamiento agresivo y los tratamientos combinados tempranos se asocian en especial con una mejora de los resultados12,13.
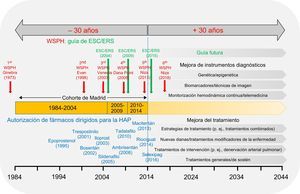
En el artículo de Quezada Loaiza et al. publicado en Revista Española de Cardiología, los autores presentan su experiencia basada en un centro de referencia nacional de HP a lo largo de un periodo de 30 años14. En un estudio observacional de cohortes, los autores describen el abordaje clínico y los factores pronósticos de 379 pacientes con HAP consecutivos que iniciaron un tratamiento dirigido entre 1984 y 2014. Además, los autores clasificaron a sus pacientes en 3 periodos de tiempo (1984-2004, 2004-2009 y 2010-2014). El fundamento para definir estos periodos fueron los avances introducidos en el tratamiento de la HAP, es decir, el desarrollo y la introducción de los tratamientos médicos dirigidos y la publicación de la guía conjunta de la Sociedad Europea de Cardiología (ESC) y la Sociedad Respiratoria Europea (ERS) para el diagnóstico y el tratamiento de la HP en 2004 y 200915,16.
En 1985, cuando se acababa de incluir a los primeros pacientes en el presente análisis, se estrenó la película de Robert Zemeckis Regreso al futuro, que llevó al cine a toda una generación de adolescentes. En esa película, Michael J. Fox (en el papel de «Marty McFly») realizaba un viaje y era transportado 30 años atrás por una máquina del tiempo. En su viaje en el tiempo hacia el pasado, «McFly» daba indicaciones para el futuro y con ello salvaba a su mentor («Doc Brown») de ser tiroteado por terroristas y evitaba que su padre fuera maltratado por su maléfico vecino «Biff». Lamentablemente, no podemos viajar al pasado y salvar a los pacientes con HAP diagnosticados a comienzos de los años ochenta aplicando las opciones de tratamiento actuales. Sin embargo, la serie de datos del centro de HP de Madrid permite examinar el impacto clínico que los avances en este campo han tenido en los pacientes tratados en un gran centro de referencia nacional. Los datos recogidos deben situarse en perspectiva en cuanto a diversos aspectos, como el desarrollo de fármacos y los conceptos de tratamiento emergentes, las recomendaciones específicas planteadas en varios simposios mundiales sobre HP y la publicación de la guía sobre HP de la ESC y la ERS (figura). De esta forma, es necesario resaltar varios factores:
- 1.
Cambio del fenotipo. Resulta muy intrigante que la media de edad de los pacientes a los que se diagnosticó HAPI en la cohorte de Madrid no cambiara con el paso del tiempo y continuara siendo de 44 años incluso en el periodo 2009-201414. Esto contrasta claramente con lo indicado por los datos de otros registros, como el COMPERA y los de Reino Unido y Francia, en que se ha observado un aumento constante de la mediana de edad al diagnóstico de los pacientes con HAPI, con medianas de 71, 62 y 55 años respectivamente, en los análisis más recientes17–19. En los pacientes ancianos, la HAPI se asoció a menudo con la presencia de múltiples factores de riesgo cardiovascular o comorbilidades, lo que ha conducido a los términos HAPI «típica» y «atípica»11,20. En el estudio AMBITION, a estos últimos pacientes se los consideró no elegibles para la inclusión en la población de análisis principal11. En general, la supervivencia de los pacientes ancianos con HAP es peor, a pesar de un menor deterioro de la hemodinámica pulmonar17, y la respuesta a los tratamientos dirigidos puede no ser la misma que la de pacientes más jóvenes. El motivo de que se observara un intervalo de edades estable en la cohorte de Madrid continúa estando en el terreno de la especulación. Los autores describen una presencia constante de HAPI y HAP hereditaria, pero con un aumento paulatino en el número de pacientes a los que se diagnosticaron formas más complejas de HP, como la enfermedad venooclusiva pulmonar (EVOP), la HAP asociada con cardiopatía congénita y la hipertensión portopulmonar (HPoP)14. El aumento de casos de EVOP puede no reflejar un aumento real de la incidencia de esta, y es posible que se explique más bien por mejor conocimiento, mayor concienciación y mejores instrumentos diagnósticos para esta entidad específica, incluidas las pruebas genéticas21. De modo análogo, el aumento relativo de la HPoP probablemente se explique por la aplicación de métodos sistemáticos de detección de HPoP, en especial antes de un trasplante de hígado4.
- 2.
Momento del diagnóstico. Se considera de importancia capital en la HAP diagnosticar e iniciar el tratamiento precozmente4. En la cohorte de España, los autores indican que un 72% de sus pacientes se encontraban en clase funcional III o IV de la OMS en el momento del diagnóstico14. Lamentablemente, esta observación concuerda con la de otros registros17–19 que indican que, incluso ahora, la mayoría de los casos de HAP se diagnostican con un retraso inaceptable. De hecho, la media de tiempo desde el inicio de los síntomas hasta el diagnóstico de la HAP que se ha descrito continúa siendo de 2,8 años. A pesar de los numerosos esfuerzos realizados para aumentar la concienciación respecto a esta enfermedad y mejorar los métodos diagnósticos, la experiencia ha resultado bastante frustrante para los médicos dedicados a la HP. Es de destacar que el porcentaje de pacientes con HAP diagnosticados en clase funcional III o IV de la OMS en el centro de Madrid mostró una disminución constante desde el 88,3% antes de 2004, hasta el 67,1% en 2004-2009 y el 58% en 2010-201414. Así pues, se debe felicitar a este centro de referencia por ser capaz de diagnosticar a los pacientes con HAP (e iniciar su tratamiento) en una fase cada vez más temprana de la enfermedad.
- 3.
Cambio de las estrategias terapéuticas. La creciente evidencia acumulada en numerosos ensayos clínicos (entre ellos los de Pulido et al.9, Sitbon et al.10 y Galiè et al.11) ha conducido a cambios sustanciales en las estrategias de tratamiento para la HAP)4,22. Mientras que las recomendaciones antiguas se basaban en monoterapia inicial, seguida de una escalada secuencial del tratamiento basada en el concepto de objetivos del tratamiento15,23, las recomendaciones actuales se decantan por un tratamiento combinado temprano o incluso inicial, con independencia de cuáles sean sus objetivos. Esto está respaldado, además, por metanálisis recientes y por los datos del ensayo AMBITION (en el que participaron solo pacientes de nuevo diagnóstico) e indica una mejora de los resultados en los pacientes con HAP en los que se utilizan tratamientos combinados12,13. Al examinar los datos del registro, queda claro que estos conceptos no son fácilmente trasladables a la práctica clínica17–20. Resulta tranquilizador observar que, en la cohorte de Madrid, el número relativo de pacientes con HAP que recibieron temprano un tratamiento combinado mostró un aumento constante, pasando del 2,3% antes de 2004 a un 10% entre 2004 y 2009 y alcanzando alrededor de un 27% entre 2010 y 201414. Este aumento de los tratamientos combinados concuerda con lo indicado por los registros actuales, pero la aplicación de las estrategias de tratamiento continúa yendo por detrás de lo recomendado en la guía y de la generación de evidencias clínicas. Será interesante observar si la frecuencia de uso de los tratamientos combinados sigue incrementándose en el futuro en este estudio observacional que continúa en marcha.
- 4.
Supervivencia. En el conjunto de su cohorte, Quezada Loaiza et al. presentan una supervivencia sin muerte ni trasplante en el primero, el tercero y el quinto año del 92,2, el 80,6 y el 68,5% respectivamente, y la mediana de supervivencia descrita fue de 9 años (intervalo de confianza del 95%, 7,532-11,959). En el subgrupo de pacientes con HAPI, la mediana de supervivencia sin muerte ni trasplante en el centro de Madrid fue de 11 años14. Estas cifras son, como mínimo, comparables a los datos recientemente publicados en los registros COMPERA, REVEAL y otros, y respaldan las mejoras que se han producido en la supervivencia en comparación con los 2,8 años observados en los pacientes con HP primaria del registro de los National Institutes of Health6,20. Es probable que esto refleje las mejoras sustanciales que se han producido en este campo con el paso del tiempo, ya que, en general, la supervivencia aumentó a medida que se incrementaban las opciones de tratamiento. Los autores determinaron también los factores pronósticos existentes en su cohorte, e indican que la supervivencia sin deterioro clínico fue mayor en las mujeres, los menores de 56 años, los pacientes con presión auricular derecha < 8mmHg, una distancia recorrida en la prueba de la marcha de 6min > 475 m, y en los que estaban en clase funcional I-II de la OMS. Esto es coherente con la estratificación del riesgo de los pacientes con HAP que recomienda la guía actual de la ESC y la ERS4. La tasa de supervivencia especialmente favorable de la cohorte de Madrid puede explicarse por el hecho de que los pacientes eran más bien jóvenes en el momento del diagnóstico, tenían una mejor clase funcional de la OMS y alcanzaron una mayor distancia recorrida en la prueba de la marcha de 6min que los pacientes de otros registros.
- 5.
Epidemia del síndrome del aceite de colza tóxico. Un aspecto peculiar de la cohorte de España es la epidemia del síndrome de aceite de colza tóxico que se produjo en este país a comienzos de la década de los ochenta. Quezada Loaiza et al. señalan que, durante esa epidemia, hubo aproximadamente 4.000 individuos afectados que contrajeron HAP, que sin embargo revirtió espontáneamente en la inmensa mayoría de los casos14. No obstante, un pequeño subgrupo de estos pacientes con el paso del tiempo contrajo una HAP grave crónica, con amplias diferencias en su periodo de latencia. Aunque se diagnosticó aproximadamente al 75% de estos pacientes antes de 2004 y constituyen el 20,3% de los pacientes con HAP del periodo 1984-2004, se siguió diagnosticando estos casos en los demás periodos y supusieron el 2,9% (2004-2009) y el 4,9% (2010-2014) del total de pacientes con HAP de nuevo diagnóstico, lo cual indica un periodo de latencia muy diverso. Esto es coherente con el concepto de que el inicio y la progresión de la HAP crónica no se inducen por un único factor, sino que requieren una «segunda agresión» o la combinación de un desencadenante con una susceptibilidad genética en pacientes específicos.
 como los World Symposia on Pulmonary Hypertension (WSPH; rojo), la guía de la Sociedad Europea de Cardiología (ESC) y la Sociedad Respiratoria Europea (ERS) sobre hipertensión pulmonar (verde), y la introducción de los tratamientos dirigidos para la HAP (azul). Se indican, además, perspectivas futuras (+ 30 años), que incluyen una mejora de los instrumentos diagnósticos y de las estrategias terapéuticas, y las futuras guías. HAP: hipertensión arterial pulmonar.")
Regreso al futuro en la HAP. Los datos recogidos en la cohorte de Madrid14 se interpretaron desde la perspectiva de los avances previos (30 años) como los World Symposia on Pulmonary Hypertension (WSPH; rojo), la guía de la Sociedad Europea de Cardiología (ESC) y la Sociedad Respiratoria Europea (ERS) sobre hipertensión pulmonar (verde), y la introducción de los tratamientos dirigidos para la HAP (azul). Se indican, además, perspectivas futuras (+ 30 años), que incluyen una mejora de los instrumentos diagnósticos y de las estrategias terapéuticas, y las futuras guías. HAP: hipertensión arterial pulmonar.
¿Qué puede deparar el futuro? Un mejor conocimiento de la genética/epigenética, el establecimiento de nuevos biomarcadores y la mejora de las técnicas de imagen deberán ayudar a detectar la HAP en estadios más tempranos y captar mejor los escenarios de alto riesgo y las respuestas al tratamiento tanto en la circulación pulmonar como en la del ventrículo derecho. Es importante señalar que, a pesar de las mejoras significativas que se han realizado, continuamos estando lejos de ser capaces de curar la HAP y los pacientes continúan sufriendo una morbilidad y una mortalidad significativas. Así pues, es crucial identificar nuevas dianas tratables y desarrollar otros tratamientos modificadores de la enfermedad adicionales. Actualmente no se considera que la HAP se deba a una pérdida de la regulación del tono vascular, sino que se reconoce como enfermedad proliferativa de los pequeños vasos de resistencia pulmonares. La investigación actual se centra en las estrategias antirremodelado, los mediadores inflamatorios y la función y el metabolismo mitocondriales7,24. ¿Es nuevo esto? La HAP fue descrita por primera vez probablemente en 1865 por Julius Klob, un anatomopatólogo austriaco, que describió la «endarteritis pulmonar deformante» como una «enfermedad que se caracteriza por un aumento de la masa de la piel vascular interna que crece y forma un tejido conjuntivo seudomembranoso»25. Resulta sorprendente e intrigante que Klob señalara también que «esta entidad, en ocasiones, aunque no siempre, se asocia con una inflamación». Así pues, lo que se observó hace más de 150 años parece continuar siendo la base de los conceptos de investigación modernos actuales. El primer paso clínico que ha permitido avanzar hacia tratamientos antirremodelado puede haber sido el ensayo clínico IMPRES, que demostró que el inhibidor de la tirosincinasa imatinib, que actúa a través de acciones antiproliferativas y proapoptóticas, mejoró sustancialmente la capacidad de ejercicio y la hemodinámica pulmonar en la HAP grave cuando se administró de manera adicional a los tratamientos dirigidos autorizados26. Aunque los motivos de preocupación en cuanto a la seguridad impidieron la autorización del imatinib para la HAP, esto puede haber tenido una importancia crucial para el seguimiento de esta estrategia prometedora y para el ulterior desarrollo de tratamientos dirigidos realmente a las alteraciones morfológicas que subyacen en las arteriolas pulmonares.
Además de la farmacoterapia moderna, también se considera importantes las medidas generales y los tratamientos de sostén4. Por ejemplo, el entrenamiento de ejercicio y los programas de rehabilitación han resultado seguros y eficaces en los pacientes con HAP, por lo que se incluyeron en las recomendaciones de la guía actual asignándoles un nivel de evidencia alto4. Sin embargo, los programas de formación estructurados son escasos, y la aplicabilidad general y la continuación fiable del ejercicio después de la fase de entrenamiento inicial continúan siendo retos importantes. Recientemente se ha introducido como nueva modalidad de ejercicio la vibración corporal total, que se realiza en una plataforma vibratoria que se mueve con oscilaciones sinusoidales, y se ha demostrado en un estudio piloto controlado que mejora la capacidad de ejercicio, la capacidad física y la calidad de vida relacionada con la salud en un plazo de 4 semanas27. Es posible que la vibración corporal total pueda llegar a incluirse en los futuros conceptos de entrenamiento estructurado y rehabilitación y brinde a estos pacientes la oportunidad de un ejercicio físico domiciliario continuo y a largo plazo.
Otro acontecimiento paralelo al inicio del estudio de Madrid (aunque más oscuro que la película de Zemeckis) es la obra de George Orwell 1984. En esa novela, que fue escrita de hecho en 1948 («el gran hermano te vigila»), Orwell intentó proyectar los desarrollos sociales que se producirían 36 años en el futuro, y en este caso produjo un cuadro oscuro de vigilancia permanente por estructuras totalitarias de las que no se podía escapar. En la medicina moderna, la monitorización de datos y la observación del estado de salud a distancia pueden considerarse beneficiosos y abiertos a una amplia variedad de aplicaciones en el futuro, como la capacidad de detectar el empeoramiento de la enfermedad de manera más temprana. Por ejemplo, recientemente se ha demostrado que la monitorización a distancia de la presión arterial pulmonar redujo sustancialmente la tasa de hospitalizaciones de los pacientes con insuficiencia cardiaca del ensayo CHAMPION28. Sin embargo, los dispositivos que permiten el seguimiento y la transmisión automática de la salud/nivel de actividad individual o de la posición geográfica a «una base de datos» pueden tener también un lado aterrador. De hecho, tendrá una importancia crucial desarrollar estos instrumentos de telemonitorización prometedores del modo más responsable posible.
Por último, una tarea importante en el futuro será mirar más allá de la HAP con objeto de comprender mejor la fisiopatología y establecer tratamientos para otras formas de HP que son mucho menos frecuentes y tienen repercusiones importantes en la supervivencia. Teniendo en cuenta el desarrollo demográfico existente en poblaciones envejecidas, el aumento de la prevalencia de la insuficiencia cardiaca con fracción de eyección conservada (IC–FEc) y la alta prevalencia de HP en esta, eso resulta especialmente cierto en el caso de la HP con IC–FEc. Aunque la enfermedad primaria objetivo se localiza claramente en el corazón izquierdo, la circulación pulmonar probablemente sea un objetivo terapéutico adicional, al menos en un subgrupo de pacientes3. Sin embargo, la investigación futura en este campo requerirá determinar detalladamente el fenotipo, incluir la caracterización hemodinámica precisa en reposo, mejorar nuestra comprensión de los cambios hemodinámicos que se producen durante el ejercicio y estudios controlados de los resultados clínicos en poblaciones de pacientes bien definidas, como los que se están llevando a cabo en la actualidad29.
Tras finalizar la obtención de los datos de la presente cohorte de Madrid, se ha publicado la guía de 2015 de la ESC y la ERS para el diagnóstico y el tratamiento de la HP4, y en 2018 se examinarán de nuevo los avances recientes en este campo y se harán nuevas recomendaciones al respecto en el sexto simposio mundial sobre HP que se celebrará en Niza (Francia). Tendrá interés observar de qué manera estas nuevas recomendaciones y los avances futuros influirán en el tratamiento de los pacientes y los resultados clínicos en el estudio observacional aún en marcha en el centro de referencia de Madrid. El futuro ha empezado ya.
CONFLICTO DE INTERESESS. Rosenkranz ha recibido pagos por conferencias y consultoría de Actelion, Bayer, GSK, Novartis, Pfizer y United Therapeutics. Su centro ha recibido subvenciones de investigación de Actelion, Bayer, MSD, Novartis y United Therapeutics. D. Dumitrescu ha recibido pagos por conferencias o consultoría de Actelion, Bayer, GSK, Novartis y Servier.