
La ausencia de compactación del ventrículo izquierdo (VI) es una anomalía estructural que se caracteriza por una capa epicárdica compactada delgada y una capa endocárdica gruesa con trabeculaciones prominentes y recesos profundos. Cuando se asocia con miocardiopatía, puede tener una causa genética. Se han descrito mutaciones en varios genes, como los de tafazzina (TAZ), alfadistrobrevina (DTNA), proteína de motivo PDZ con corte y empalme alternativo de banda Z (ZASP), lamina A/C (LMNA) y genes que codifican proteínas sarcoméricas1. Sin embargo, el rendimiento de las pruebas genéticas parece ser relativamente bajo y permite identificar variantes de interés clínico en aproximadamente un 40% de los casos. Con frecuencia, la escasa información disponible sobre los genes involucrados dificulta la clasificación de las variantes identificadas2. La mayor parte de los estudios no han evaluado la posible presencia de variaciones en el número de copias (CNV). Las CNV, que se definen como ganancias o pérdidas de ADN, contribuyen a la aparición de múltiples trastornos genéticos y pueden ser causa de rasgos complejos. Concretamente, la deleción de 1p36 se ha asociado con la aparición de miocardiopatía no compactada del VI en el contexto de un fenotipo característico3. Arndt et al. señalaron que las manifestaciones cardiacas probablemente dependan de la alteración del gen PRDM16. El análisis de cobertura obtenida mediante secuenciación de nueva generación (NGS) permite la adecuada detección de las CNV. Por esta razón, se incluyó el gen PRDM16 en nuestros paneles de enfermedades cardiovasculares hereditarias y se evaluó sistemáticamente la presencia en nuestros pacientes de CNV en este gen. Se identificaron 2 deleciones de PRDM16 en un total de 382 pacientes remitidos por sospecha clínica de miocardiopatía no compactada del VI. No se identificó ninguna deleción de este gen en más de 12.000 pacientes remitidos que tenían otros fenotipos vasculares.
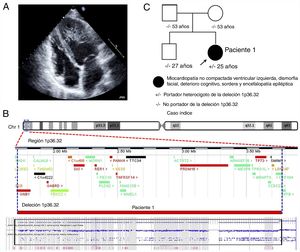
La paciente 1 era una mujer de 25 años con una miocardiopatía no compactada del VI (figura 1A) y un fenotipo complejo, diagnosticada a la edad de 6 meses, con dismorfia facial leve, deterioro cognitivo y encefalopatía epiléptica. Se descartó la presencia de sordera neurosensorial. No se pudo realizar una cardiorresonancia magnética debido a claustrofobia, y el registro Holter no mostró arritmias. Sus padres y su hermano no estaban afectados. El estudio de CNV basado en el análisis de cobertura obtenida mediante NGS identificó una deleción heterocigota del gen PRDM16. Un array de polimorfismos de nucleótido único (SNP) confirmó la deleción de una región formada por 112 genes, que incluía el gen PRDM16 (figura 1B). Se confirmó que se trataba de una deleción aparecida de novo (figura 1C) y se la consideró patogénica.
. C: el estudio de segregación familiar mediante chip de SNP confirmó que esta deleción había aparecido de novo. CNV: variaciones en el número de copias. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Paciente 1. A: ecocardiografía en la que se observa una hipertrabeculación del ventrículo izquierdo medioapical prominente con una disfunción sistólica moderada. B: el análisis de CNV mediante un estudio de microchip de SNP confirmó la deleción de 112 genes, incluido PRDM16 (Affymetrix CytoScan). C: el estudio de segregación familiar mediante chip de SNP confirmó que esta deleción había aparecido de novo. CNV: variaciones en el número de copias. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
El paciente 2 era un varón de 23 años evaluado tras la muerte súbita de su padre a la edad de 43 años. En este caso, no se realizó autopsia. La ecocardiografía mostró ausencia de compactación del VI, sin deterioro de la función sistólica (figura 2A). La cardiorresonancia magnética no mostró realce tardío de gadolinio y no se detectaron arritmias en el registro Holter. No se observó ninguna anomalía extracardiaca. El estudio de CNV basado en el análisis de cobertura obtenida mediante NGS identificó una deleción heterocigota de los exones 2 a 17 del gen PRDM16 en el probando y confirmó también esta deleción en su hermana de 20 años. Esta presentaba ausencia de compactación del VI y disfunción sistólica leve (figura 2B). Un array de SNP realizado en el probando confirmó la deleción de 11 genes, entre los que se encontraba PRDM16 (figura 2C). La deleción no se daba en la madre no afectada (figura 2D) y se la consideró probablemente patogénica. Sin embargo, no se pudo evaluar la presencia de la mutación en el lado paterno.
. D: el estudio de segregación familiar mediante NGS confirmó la presencia de esta deleción en la hermana. CNV: variaciones en el número de copias; VI: ventrículo izquierdo. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Paciente 2. A: ecocardiografía que muestra la ausencia de compactación en los segmentos apical y lateral del VI. B: ausencia de compactación en el VI de la hermana afectada. C: el análisis de CNV mediante un estudio de microchip de SNP confirmó la deleción de 11 genes, incluido PRDM16 (Affymetrix CytoScan). D: el estudio de segregación familiar mediante NGS confirmó la presencia de esta deleción en la hermana. CNV: variaciones en el número de copias; VI: ventrículo izquierdo. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
La deleción de 1p36 heterocigota es el síndrome de deleción subtelomérica más frecuente. Se cree que afecta a entre 1/5.000 y 1/10.000 recién nacidos, aunque es posible que sean subestimaciones. El fenotipo asociado se caracteriza por retraso psicomotor, déficits auditivos, crisis epilépticas, características faciales dismórficas, miocardiopatía no compactada/dilatada, discapacidad intelectual y otras anomalías congénitas4. Hay una gran variabilidad clínica entre los distintos individuos, que podría explicarse por las diferencias en la longitud de las deleciones o en los genes afectados. Algunos de esos genes se han asociado de manera individual con defectos cardiacos y anomalías del desarrollo5. Arndt et al.3 utilizaron la hibridación in situ para identificar una región de deleción frecuente en pacientes con un síndrome de deleción de 1p36 que presentaban miocardiopatía. Esta región incluía tan solo los 14 exones terminales del gen PRDM16. Estos autores observaron también variantes patogénicas en algunos casos de miocardiopatía no compactada no sindrómica y dilatada. Se han descrito otras anomalías cromosómicas (p. ej., deleciones de 1q43, 5q35 y 8p23.1) en ausencia de compactación del VI sindrómica6.
Se presentan 2 nuevos casos de miocardiopatía no compactada del VI asociada con síndrome de deleción de 1p36 en una cohorte amplia de pacientes con sospecha de la enfermedad, uno de ellos con una forma de presentación no sindrómica que pone de manifiesto la utilidad de la NGS en la identificación de la causa genética de la enfermedad. El estudio genético con análisis de CNV debería incluirse en el protocolo diagnóstico de la miocardiopatía no compactada del VI. Esto podría llevar también a la identificación de nuevos genes candidatos incluidos en las regiones de CNV.
En conclusión, la evaluación sistemática de CNV en el estudio genético mediante NGS puede ser útil para el estudio de pacientes con miocardiopatía no compactada del VI. Por lo general son necesarios métodos complementarios para confirmar/caracterizar este tipo de variantes. Esta estrategia podría aumentar el rendimiento diagnóstico en estos pacientes y limitar el porcentaje de falsos negativos. Estos datos son de gran importancia para el asesoramiento genético adecuado y los exámenes de detección sistemática en las familias de los pacientes.
FINANCIACIÓNLos autores no recibieron ninguna financiación específica para este trabajo.
CONFLICTO DE INTERESESJ.P. Trujillo-Quintero, D. De Uña-Iglesias y L. Monserrat forman parte de la compañía de diagnóstico genético Health in Code.