Palabras clave
INTRODUCCIÓN
La enfermedad cardiovascular (ECV) de etiología aterotrombótica es la primera causa de muerte en el mundo occidental1,2. Aunque España tiene una incidencia menor de ECV que los países del norte de Europa3, debemos tener en cuenta que los ingresos a causa de síndrome coronario agudo (SCA) sufren un incremento anual del 1,5% y nos estamos acercando a las cifras europeas4.
Nuestro grupo ha revisado recientemente en esta misma Revista5 las bases bioquímicas del papel protector del colesterol de las lipoproteínas de alta densidad (cHDL). Por ello, aquí nos centraremos, de una manera más específica, en las evidencias que apoyan el papel protector del cHDL en la ECV y en las diversas posibilidades para incrementar sus concentraciones.
¿PODEMOS MEJORAR LA PREVENCIÓN CARDIOVASCULAR QUE OFRECEN LAS ESTATINAS?
La reducción de las concentraciones de colesterol asociado a lipoproteínas de baja densidad (cLDL) mediante estatinas produce un descenso muy significativo del riesgo cardiovascular. Se produjo un cambio de paradigma en el tratamiento de la ECV con la publicación del estudio 4S, que mostraba una reducción de la mortalidad total del 29% en pacientes isquémicos tratados con simvastatina6. Desde entonces, numerosos estudios han confirmado que las estatinas reducen el riesgo cardiovascular un 20-30%7-9. De hecho, un metaanálisis reciente (14 ensayos clínicos con estatinas, 90.056 pacientes, media de seguimiento de 5 años) concluyó que, por cada 40 mg/dl que disminuya el cLDL, se reducen hasta un 21% los eventos cardiovasculares10. Estas observaciones apuntaban a la posibilidad de que reducciones más intensas de cLDL podrían inducir mayores beneficios. De hecho, esta hipótesis se confirmó en estudios que emplearon dosis mayores o estatinas más potentes, como IDEAL11, TNT (Treating to New Targets)12 y PROVE-IT8 (fig. 1)13. Estas evidencias llevaron a postular el paradigma «the lower, the better» (cuanto más bajos los valores de cLDL, mejor), lo que indica la necesidad de una reducción más agresiva de las concentraciones de cLDL. El estudio ASTEROID apuntó la posibilidad de que unos valores de 60 mg/dl de cLDL podían incluso llegar a reducir lesiones coronarias previamente establecidas14. Todas estas observaciones indican la necesidad de obtener unos valores de cLDL mucho más bajos que los recomendados en la actualidad.

Fig. 1. Reducción de eventos cardiovasculares (objetivo de valoración clínico) mediante tratamiento con estatinas. Estudios con: rojo, atorvastatina; azul, simvastatina; verde, pravastatina; rosa, lovastatina. Modificado, con permiso, de Ibañez et al13.
El principal problema en la aplicación masiva de este paradigma radica en la dificultad de alcanzar unas concentraciones plasmáticas tan reducidas. Este inconveniente está claramente representado por los resultados de dos estudios, uno en Europa15 y otro en Estados Unidos16, que muestran que menos del 50% de los pacientes consiguen esos objetivos. Para agravar la situación, el estudio L-TAP2 indicaba que los pacientes con mayor riesgo eran los que en menor proporción alcanzaban los valores deseados de cLDL17. En segundo lugar, el riesgo residual de sufrir otro evento cardiovascular se mantiene muy elevado, incluso a pesar de tratamientos intensos con estatinas. Por ejemplo, pese a conseguir valores de cLDL de 62 mg/dl en el estudio PROVE-IT8, el riesgo residual de muerte y evento cardiovascular en pacientes isquémicos se mantenía elevado (hasta el 22,4% tras 2 años de seguimiento). Es decir, si conseguimos reducir el riesgo cardiovascular en un 20-30%, esto significa que hay un 70-80% de eventos cardiovasculares que no podemos reducir, una cifra inaceptablemente elevada. Por este motivo, la comunidad cardiológica está centrando su atención en reducir dicho riesgo residual mediante otras estrategias distintas y complementarias a las estatinas, como el incremento de las concentraciones de colesterol de las lipoproteínas de alta densidad (cHDL).
En conclusión, todas las evidencias apoyan que, además de la adopción de un estilo de vida más saludable, las estatinas sean la primera elección en el tratamiento de pacientes con ECV. No obstante, a pesar del tratamiento con estatinas, todavía es posible mejorar las posibilidades terapéuticas, reducir la placa18 y reducir el impacto socioeconómico de esta enfermedad mediante estrategias adicionales, como el incremento del cHDL.
¿POR QUÉ CENTRARNOS EN ESTUDIAR EL cHDL?
Evidencias epidemiológicas
Numerosos estudios epidemiológicos han demostrado una correlación inversa entre los valores de cHDL y el riesgo cardiovascular19-26 (tabla 1). Las primeras observaciones epidemiológicas se realizaron en 1950 en esquimales27 y posteriormente fueron corroboradas en 1975 por los hermanos Miller28. Apenas 2 años más tarde, los estudios de Tromsø19 y de Framingham29 confirmaron prospectivamente que una baja concentración de cHDL predecía futuros episodios cardiovasculares, y era —junto con la relación colesterol total (CT)/cHDL— el único predictor independiente de ECV. Modernos estudios (PROCAM23 en Alemania, Goldbourt24 en Israel) ratifican esta relación; los individuos con cHDL < 35 mg/dl tienen una incidencia de eventos cardiovasculares 8 veces mayor que los individuos con cHDL > 65 mg/dl20. Un metaanálisis30 de cuatro estudios poblacionales previos (FHS29, LRCF31, CPPT22 y MRFIT21) mostró que por cada incremento de 1 mg/dl en el cHDL se producía una reducción de riesgo cardiovascular del 1,9-2,3% en los varones y del 3,2% en las mujeres. Esta relación se mantiene incluso ante valores de cLDL bajos32. En un estudio post hoc del ensayo clínico TNT, incluso en pacientes con valores de cLDL inferiores a 70 mg/dl, aquellos en el quintil superior de cHDL presentaban un riesgo de eventos cardiovasculares menor que los situados en el quintil inferior (p = 0,03).

Finalmente, estudios recientes confirman estos datos clásicos. Un nuevo subanálisis del estudio Framingham desde 1975 hasta 200333 muestra una reducción del 21% en el riesgo cardiovascular por cada 5 mg/dl de elevación del cHDL. Se observó una interacción muy importante: cuanto menores eran los valores de cLDL, mayor era el impacto protector de elevar las concentraciones de cHDL. Se trata de una observación de importancia capital en un momento como el actual, en que las guías recomiendan valores de cLDL < 70 mg/dl en pacientes isquémicos o diabéticos. Además, un estudio prospectivo confirma que, en pacientes ancianos (> 85 años), los valores bajos de cHDL y no los elevados de cLDL eran predictores de mortalidad cardiovascular34. Un subanálisis del ensayo clínico MIRACL35 demuestra que un cHDL bajo predice el riesgo de eventos cardiovasculares recurrentes a corto plazo (4 meses) tras sufrir un SCA, mientras que los valores altos de cLDL no parecen ser un factor pronóstico. Para concluir, un reciente metaanálisis36 de cuatro ensayos clínicos prospectivos que usaron IVUS (REVERSAL, CAMELOT, ACTIVATE, ASTEROID) demostró que, para detener la progresión de la placa o conseguir su reducción, se precisan simultáneamente tanto unas concentraciones de cLDL < 87,5 mg/dl como una elevación > 7,5% en los valores de cHDL.
Esta relación inversa entre cHDL y riesgo cardiovascular adquiere radical importancia al constatar la enorme prevalencia de las concentraciones bajas de cHDL en la población; se trata de la alteración del perfil lipídico más frecuente en la ECV prematura37. En un reciente estudio sobre pacientes (n = 231.986) hospitalizados con ECV38, aproximadamente la mitad de ellos tenía concentraciones bajas de cLDL(< 100 mg/dl), pero el 50% tenía un cHDL < 40 mg/dl y únicamente un 10% tenía un cHDL > 60 mg/dl. Asimismo, es un componente por definición del síndrome metabólico (que tiene una prevalencia del 23,7% entre los mayores de 20 años en Estados Unidos)39. Todas estas observaciones apoyan claramente los beneficios clínicos derivados de un tratamiento conjunto de los valores de cLDL y cHDL.
Evidencias preclínicas
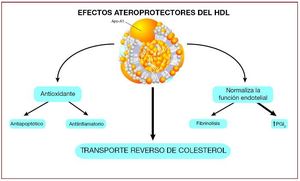
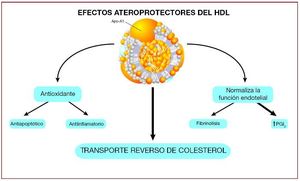
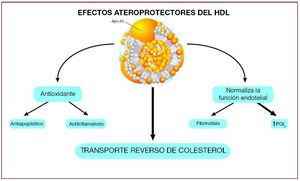
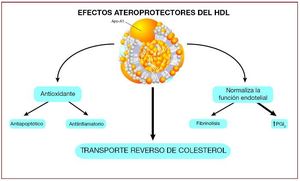
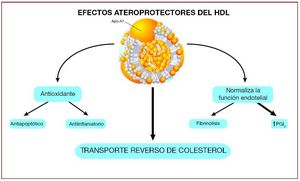
En 1968 Glomset postuló que el cHDL facilita el retorno del colesterol desde los tejidos periféricos hasta el hígado, para allí ser eliminado a través de la bilis y las heces40, un proceso que denominó transporte reverso de colesterol (TRC). En 1973 Ross y Glomset propusieron que el TRC podía ser un mecanismo protector frente a la aterosclerosis41. No obstante, no había datos experimentales sólidos que confirmaran las evidencias epidemiológicas y el tema fue objeto de controversia durante varios años. Nuestro grupo fue el primero en demostrar en un modelo experimental de aterosclerosis de conejo que la administración de cHDL no sólo produce inhibición, sino que consigue regresión de la lesión aterosclerótica y su contenido lipídico42,43. Esta hipótesis se confirmó con animales transgénicos, en los que la sobreexpresión de apo-A1 (la principal apolipoproteína componente del cHDL) inhibe la progresión de aterosclerosis, e incluso consigue la regresión de aterosclerosis en ratones44,45. De manera inversa, los ratones knock-out para apo-A1 desarrollan aterosclerosis de manera más grave y acelerada que los wild-type46. Se ha demostrado recientemente que el cHDL tiene efectos beneficiosos diferentes del TRC (fig. 2); dado que este tema escapa al objetivo del presente artículo, remitimos al lector a excelentes revisiones al respecto47.

Fig. 2. Efectos ateroprotectores del colesterol de las lipoproteínas de alta densidad (cHDL). Modernos estudios apuntan a que los efectos beneficiosos del cHDL no solamente están mediados por el transporte reverso del colestorol, sino también por sus efectos antioxidantes, antiagregantes, antiinflamatorios y de mejora de la función endotelial47.
Evidencias clínicas
Se han llevado a cabo numerosos ensayos clínicos para constatar los efectos de la elevación del cHDL mediante diversos fármacos. La tabla 2 ofrece un resumen sucinto de ellos sin ánimo exhaustivo48-86.


ESTRATEGIAS PARA ELEVAR LAS CONCENTRACIONES DE cHDL
Así pues, la doble estrategia de disminuir las concentraciones de cLDL e incrementar las de cHDL es la idónea en la prevención de ECV (fig. 3). ¿Por qué no nos hemos centrado hasta ahora en subir el cHDL? Básicamente porque no teníamos fármacos capaces de subir el cHDL sin efectos adversos, sólo medidas no farmacológicas y un medicamento eficaz como la niacina pero con graves problemas de tolerabilidad. A continuación revisaremos someramente las terapias disponibles en la actualidad para elevar los valores de cHDL.

Fig. 3. Tratamiento óptimo para la aterosclerosis. Reducir las concentraciones de colesterol de las lipoproteínas de baja densidad (cLDL) o subir las de colesterol de las lipoproteínas de alta densidad (cHDL) desplaza el fiel de la balanza hacia la «regresión» de la lesión. Por ello, el mejor tratamiento contra la enfermedad arteriosclerótica es la combinación de ambas estrategias.
Medidas no farmacológicas
La tabla 3 resume los efectos beneficiosos de las siguientes medidas:

- El ejercicio aeróbico frecuente aumenta el cHDL aproximadamente un 5%87-89. Este efecto es precoz (en menos de 2 meses)86 y parece ligado a la frecuencia, la intensidad y la duración del ejercicio.
- Abandono del hábito tabáquico: incrementa los valores de cHDL en 5 mg/dl90,91, incluso en plazos tan cortos como 2 semanas después del cese.
- Pérdida de peso: un reciente metaanálisis ha demostrado que en pacientes obesos la pérdida de cada kilogramo de peso se asocia a un incremento del cHDL de 0,35 mg/dl92.
- El consumo de cantidades moderadas de alcohol (30-40 g diarios; se recomienda 2 bebidas en varones y 1 en mujeres) incrementa las concentraciones de cHDL un 5-15% y disminuye el riesgo cardiovascular93-95. Aparentemente el alcohol etílico per se causa el ascenso, por lo que cualquier bebida alcohólica podría elevarlo95. No obstante, los beneficios deben sopesarse con los riesgos de su consumo antes de recomendar la ingesta de alcohol.
- Factores dietéticos: las dietas ricas en ácidos grasos monoinsaturados y poliinsaturados (pescado azul, frutos secos, aceite de oliva) elevan los valores de cHDL y reducen el riesgo cardiovascular96. El consumo de ácidos grasos saturados reduce el potencial antiinflamatorio del cHDL, mientras que los ácidos poliinsaturados mejoran dicho potencial97.
Medidas farmacológicas «clásicas»
Estatinas
Las estatinas elevan el cHDL un 5-10%86 (la rosuvastatina es la que induce unos incrementos mayores de cHDL98) al aumentar la síntesis de apo-A199 y disminuir la actividad de la proteína de transferencia de ésteres de colesterol (CETP). Los efectos de las estatinas en el cHDL dependen de los valores iniciales de éste; se obtiene un efecto más marcado cuanto menores son los valores iniciales del cHDL.
Niacina/ácido nicotínico
La niacina fue el primer fármaco hipolipemiante que demostró un efecto favorable en los lípidos plasmáticos (Altschul la estudió ya en los años cincuenta) y el primero en disminuir el tamaño de los xantomas tendinosos y reducir las complicaciones cardiovasculares de la aterosclerosis.
La niacina reduce la captación del cHDL por el hígado (holopartícula)5,100 y la cantidad de apo-A1 extraída, lo que da lugar a partículas de cHDL ricas en apo-A1 (muy eficientes en el TRC). También reduce la actividad de la CETP y la lipolisis y la liberación de ácidos grasos hacia el hígado, con la consiguiente disminución en la producción de lipoproteínas de muy baja densidad (VLDL). Es el tratamiento más efectivo para elevar el cHDL (20-35%); reduce el CT un 10-15%, el cLDL un 15-20%, los triglicéridos (TG) un 30-50% y es el único que reduce la Lp(a) (29-35%).
Varios ensayos clínicos han demostrado que la niacina reduce uniformemente los eventos cardiovasculares y la progresión de la aterosclerosis. Los resultados de los ensayos clínicos con niacina se resumen en la tabla 2. Para confirmar la hipótesis de si añadir niacina a pacientes isquémicos con cHDL bajo ya tratados con estatinas reduce los eventos cardiovasculares clínicos, se está realizando el ensayo clínico AIM-HIGH (se espera que finalice en 2011). La combinación de estatinas y niacina sí ha demostrado mejorar los objetivos de valoración de imagen, tanto en los estudios ARBITER 360 y 661 como en un reciente estudio en que reduce las placas de ateroma carotídeas evaluadas mediante resonancia magnética62.
La niacina tiene una alta tasa de efectos secundarios. El fundamental es el flushing o sofoco (hasta en el 80% de los pacientes), una combinación de rubor, calor y picor que empieza en la cara y puede extenderse a brazos y cuerpo; suele empezar entre 30 y 120 min tras la administración del fármaco y dura aproximadamente media hora. La frecuencia y la intensidad del sofoco llegan a afectar en gran manera el cumplimiento terapéutico. Se puede minimizar con una titulación lenta de la dosis de niacina, tomando aspirina media hora antes, evitando las comidas picantes o calientes y tomando simultáneamente algo de comida baja en calorías.
Fibratos
Son agonistas de los receptores activados de proliferación de peroxisomas alfa (PPARα). Incrementan la expresión de apo-A1, apo-A2 y lipoproteinlipasa y reducen la apo-C3 y la actividad de la proteína de transferencia de ésteres de colesterol (CETP)101. Reducen los valores de VLDL al aumentar la oxidación de ácidos grasos en el hígado, reducir la lipogénesis y favorecer la captación de ácidos grasos por el músculo. Aumentan la concentración de cHDL un 10-20% y reducen los valores de TG un 20-50% y de cLDL un 10-15%. El fenofibrato y el bezafibrato diminuyen más el cLDL, mientras que el gemfibrozilo reduce más los TG. Su impacto depende de los valores lipídicos basales; las elevaciones de cHDL son más marcadas cuando las concentraciones basales de TG están elevadas que cuando están bajas101.
En estudios de imagen (objetivo indirecto), tanto el gemfibrozilo74 y el bezafibrato73, en sujetos con cHDL bajo, como el fenofibrato, en sujetos con diabetes mellitus tipo 2 (DM2)75, han demostrado reducir la progresión de arteriosclerosis coronaria.
Sin embargo, sí parece haber diferencias entre los fibratos en los estudios con objetivos de valoración clínica. Dos estudios (Helsinki Heart Study68 y VA-HIT69) demuestran que el gemfibrozilo reduce significativamente los eventos cardiovasculares; sin embargo, el bezafibrato70,71 y el fenofibrato72 no han demostrado una disminución del riesgo cardiovascular e incluso hay diversos estudios que muestran que el clofibrato aumenta la mortalidad general67.
Tiazolidinedionas
Indicadas en el tratamiento de la DM2, son agonistas de los PPARγ que actúan aumentando la sensiblidad a la insulina en el tejido graso y el hígado. Favorecen la captación de glucosa y disminuyen tanto su producción hepática como la concentración de ácidos grasos libres circulantes. Tanto la pioglitazona como la rosiglitazona han demostrado una actividad hipoglucemiante similar (ambas disminuyen la hemoglobina glucosilada un 1,5%); no obstante, la pioglitazona es superior en cuanto a los efectos cardiovasculares tanto por su perfil lipídico (incrementa el cHDL un 10%, reduce en mayor cuantía los TG y no altera el cLDL; mientras que la rosiglitazona eleva el cLDL un 10%102) como por los resultados de los estudios. Recientemente se han publicado dos ensayos clínicos con efectos positivos en objetivos de valoración de imagen: la pioglitazona en PERISCOPE82 retrasó la progresión de la aterosclerosis coronaria (medida por IVUS) y en CHICAGO85 redujo la progresión del grosor íntima-media (GIM) carotídeo. En el estudio PROACTIVE84 con 5.238 sujetos con DM2, la pioglitazona no redujo el objetivo primario clínico (muerte, infarto agudo de miocardio [IAM], accidente cerebrovascular [ACV], necesidad de revascularización o de amputación de extremidades inferiores), pero sí el objetivo secundario especificado a priori (muerte, IAM, ACV) en un 16%. PROACTIVE ha sido muy criticado por incluir en su objetivo primario revascularizaciones y amputaciones (cuya indicación puede variar según los centros).
Los efectos secundarios de las glitazonas son ampliamente conocidos, como el incremento del riesgo de fracturas y de retención hídrica en el organismo (pueden desencadenar insuficiencia cardiaca). El metaanálisis de Lincoff103 sobre la pioglitazona, con 19 estudios y 16.390 pacientes, fue el primero en mostrar que si bien parece reducir el riesgo cardiovascular en un 18%, asimismo aumentaba el riesgo de insuficiencia cardiaca (ICC) grave. Por su parte, la rosiglitazona no sólo produce ICC, sino que aumenta el riesgo de IAM104,105.
Por otra parte, el rimonabant era el primer inhibidor selectivo del receptor cannabinoide tipo 1 (localizado principalmente en el hipotálamo pero también en los músculos, el tejido graso y el aparato digestivo). Merced a sus marcadas propiedades anorexígenas, conseguía descensos de peso del 5% (tras restar el efecto de la dieta), de perímetro abdominal de hasta 4 cm y de grasa subcutánea. Respecto al perfil lipídico, carecía de efecto en el cLDL, pero incrementaba el cHDL un 10% y reducía los TG un 15%106-108, sólo un 50% de estas variaciones lipídicas era debido a la pérdida de peso. No obstante, en el estudio STRADIVARIUS83 no modificó el GIM carotídeo. Dados sus efectos psiquiátricos adversos (depresión, ansiedad y riesgo de suicidio)109, se ha retirado del mercado.
En la tabla 4 se resumen los principales efectos secundarios de estos fármacos.

Nuevas posibilidades
Niacina de liberación prolongada/laropiprant
La unión de la niacina al receptor GPR109A en las células de Langerhans de la piel libera prostaglandina D2 (PGD2), cuyos efectos vasodilatadores causan flushing. La adición de laropiprant (inhibidor de DP1, el receptor de PGD2) a la niacina de liberación prolongada reduce el flushing hasta un 74% sin afectar al perfil lipídico110. Dicha combinación está aprobada en Europa, pero no en Estados Unidos, pues la FDA está esperando los resultados del ensayo clínico HPS2-THRIVE (se esperan para 2013) para comprobar si la adición de esta combinación al tratamiento estándar con estatinas reduce los eventos cardiovasculares en pacientes isquémicos.
Inhibidores de la CETP
La CETP cataliza la transferencia de ésteres de colesterol (EC) de HDL a LDL-VLDL a cambio de TG100. En Japón, en 1989, se describió a individuos con cifras extraordinariamente elevadas de cHDL debido a una baja actividad de la CETP111. Esta estrategia aparentemente tan prometedora de incrementar el cHDL mediante la inhibición farmacológica de la CETP se vio inesperadamente truncada cuando, en el estudio ILLUMINATE76, el torcetrapib mostró un incremento significativo de eventos cardiovasculares y de mortalidad, pese a incrementos de cHDL del 72% y descensos de cLDL del 25%. Los estudios de imagen demostraron resultados muy concordantes: tanto IVUS (ILLUSTRATE77) como GIM carotídeo (RADIANCE 178 y RADIANCE 279) mostraron tasas similares de aterosclerosis entre torcetrapib y placebo; de hecho, los objetivos secundarios mostraron progresión de aterosclerosis con torcetrapib. A pesar del fracaso del torcetrapib, hay otros dos inhibidores de la CETP, el anacetrapib112 y el dalcetrapib113, actualmente en desarrollo, con buenos resultados.
Existen dos hipótesis plausibles para explicar el fracaso del torcetrapib:
- Efectos off-target: el torcetrapib per se tiene un efecto tóxico no relacionado con la inhibición de la CETP, pues eleva la presión arterial media 4,6 mmHg (en algunos pacientes hasta 15 mmHg) e incrementa las concentraciones de sodio, bicarbonato y aldosterona, efectos que parecen ser debidos a la toxicidad suprarrenal y la activación del sistema angiotensina-aldosterona76. De hecho, el anacetrapib y el dalcetrapib incrementan el cHDL un 139% sin afectar a la presión arterial.
- Generación de partículas de cHDL ineficaces. Significado de las cifras de cHDL frente a valores de apo-A1. Una de las hipótesis usadas para explicar el fracaso del torcetrapib es la posibilidad de que este agente generase una partícula de HDL ineficaz para el TRC a pesar de los elevados valores de cHDL que presentaban estos pacientes. La gran importancia entre la calidad y la cantidad de las partículas de cHDL se representa en la figura 4, que presenta la síntesis y la maduración metabólica de la partícula de cHDL100. Las partículas de cHDL naciente contienen proporcionalmente mucha apo-A1 y poco colesterol; por lo tanto, son muy efectivas para el TRC. Mediante su maduración metabólica, la partícula de cHDL incrementa su contenido en lípidos (EC) y se convierte en una partícula más esférica. El tratamiento con torcetrapib causa un aumento significativo de partículas grandes y maduras de cHDL, sobrecargadas de EC, con escasa capacidad para promover la salida de colesterol de los macrófagos; es decir, se incrementa la cantidad de colesterol transportado por la fracción de HDL (el cHDL medido en los análisis clínicos sistemáticos), pero no su capacidad removedora de colesterol de las estructuras extrahepáticas (verbigracia, los macrófagos); explicado de otra manera, aumentan los valores de cHDL (cantidad), pero no se eleva su capacidad removedora (calidad) (no se incrementa el TRC, los valores de apo-A1 permanecen iguales, como se puede apreciar en la fig. 5).

Fig. 4. Metabolismo del colesterol asociado a las lipoproteínas de alta densidad (cHDL) y el transporte reverso de colesterol (TRC). Apo-A1 se sintetiza en el hígado y el intestino donde, a través del transportador ABCA-1, recibe una pequeña cantidad de fosfolípidos (FL) y se transforma en apo-A1 pobre en lípidos. En la circulación periférica recibe colesterol libre (CL) a través de ABCA-1 (HDL naciente, con migración preβ1). Mediante la acción de la enzima LCAT, el CL pasa a ésteres de colesterol (EC), y así se transforma en cHDL maduro esférico (HDL3 y HDL2). Dicho cHDL maduro recibe colesterol de los tejidos periféricos a través de SR-B1 o de ABCG1, aumentando su tamaño y su contenido de EC. El TRC se completa por dos vías: a) captación hepática de cHDL maduro a través de SR-B1, y b) la CETP cataliza la transferencia de EC a colesterol asociado a las lipoproteínas de baja densidad (cLDL), los cuales a su vez serán captados por el hígado a través del receptor para LDL (LDL-R). Finalmente, desde el hígado el colesterol se excreta por la bilis al intestino.

Fig. 5. Cantidad frente a calidad de colesterol de las lipoproteínas de alta densidad (cHDL). Esta figura representa la maduracion metabólica de las HDL. Nótese que una misma particula de HDL puede dar unos valores diferentes de cHDL. La HDL naciente (inmadura) contiene en proporción relativa, mucha apo-A1 (en este ejemplo, 1 unidad arbitraria) y poco colesterol (en este ejemplo, 1). Conforme va madurando, va acumulando cada vez más colesterol (50 unidades para HDL2, en este ejemplo), manteniendo idéntica la cantidad de apo-A1 (1 unidad). Cuando en la clínica medimos los valores de HDL, estamos midiendo cHDL total (en el ejemplo, para HDL2 sería 50). No obstante, el torcetrapib genera partículas grandes de HDL, sobrecargadas de colesterol (es decir, en el análisis ofrecen cifras muy elevadas de cHDL), pero ineficaces para el transporte reverso de colesterol (pues los valores de apo-A1 se mantienen iguales). Esta observación es la que se ha barajado para justificar los resultados del estudio ILLUMINATE, donde hubo un incremento en la mortalidad a pesar de generarse unos valores elevados de cHDL.
Los estudios epidemiológicos y genéticos ofrecen asimismo resultados contradictorios. Un subestudio de Framingham (1.978 participantes, seguimiento de 15 años) indica que una actividad reducida de la CETP conlleva un riesgo elevado de ECV114. De manera similar, el estudio LURIC (3.256 pacientes remitidos a coronariografía, seguimiento de 7,75 años) relaciona los valores bajos de CETP con una mayor mortalidad115. Por el contrario, un estudio con más de 70.000 sujetos llega a la conclusión opuesta; las variaciones genéticas (SNP) en el gen de la CETP alteran el perfil lipídico, pero no incrementan la presión arterial, lo cual indica que los efectos deletéreos del torcetrapib fueron off-target y que otros inhibidores de la CETP no tienen por qué producirlos. No obstante, todos estos estudios discordantes arrojan dudas sobre la utilidad de la inhibición de la CETP.
Agonistas de receptores LXR
Los LXR actúan como factores nucleares de transcripción que se asocian con los receptores retinoides X (RXR), forman heterodímeros e inducen la expresión de determinados genes116. El LXRα se expresa con muy alta densidad en el hígado y en valores más bajos en las suprarrenales, el intestino, el tejido adiposo, los macrófagos, los pulmones y el riñón; mientras que el LXRβ es universalmente expresado (la isoforma predominante en el hígado es LXRα).
Los agonistas del LXR aumentan el TRC116 (fig. 4) al incrementar la expresión de ABCA1 (transporta colesterol de macrófagos a HDL inmaduro), ABCG1 (transporta colesterol de macrófagos a HDL maduro), ABCG5/ABCG8 (excreta colesterol desde el hígado a la bilis) y 7-α-hidroxilasa (enzima limitante en la síntesis de ácidos biliares). Asimismo, mejoran la tolerancia a la glucosa en modelos animales y tienen propiedades antiapoptóticas y antiinflamatorias.
Los agonistas sintéticos del LXR reducen la aterosclerosis en modelos murinos117. No obstante, entre sus efectos secundarios destacan la hipertrigliceridemia y la esteatosis hepática (debido a la inducción de la molécula hepática SREBP-1c)118. Existen dos alternativas para evitar estos efectos secundarios, bien moduladores de LXR selectivos para tejidos específicos (los macrófagos más que el hígado), bien desarrollar agonistas del LXR selectivos para cada isoforma (dado que el LXRβ parece inducir el transporte reverso de colesterol sin las complicaciones hepáticas atribuidas al LXRα). De hecho, se ha descrito que un agonista selectivo del LXR induce menos esteatosis hepática119.
Apo-A1 Milano
En 1985 Sirtori y Franceschini estudiaron un pequeño grupo de personas en la pequeña villa italiana de Limone sul Garda, a orillas del lago Como, que compartía un perfil lipídico con concentraciones extremadamente bajas de cHDL y apo-A1, valores elevados de TG y, sorprendentemente, un riesgo cardiovascular muy bajo120. Se identificó que este perfil era causado por una mutación (sustitución de la arginina por cisteína en posición 173) en el gen de apo-A1; dada la baja tasa de eventos cardiovasculares, se postuló la teoría de que esta molécula, denominada apo-A1Milano, es mucho más funcional que la apo-A1 wild-type121.
Las administraciones repetidas del complejo apo-A1Milano recombinante/fosfolípidos (ETC-216) consiguen una regresión de la aterosclerosis en ratones122. Nuestro grupo ha demostrado en el conejo que el ETC-216 produce regresión (de un 4,2% en volumen de placa, medida por resonancia magnética) y estabilización de la placa (reducción en infiltración de macrófagos y en expresión de factor tisular, MCP-1, COX2 y MMP2)123.
Estos hallazgos se han confirmado en humanos usando el complejo apo-A1Milano recombinante con fosfolípidos (ETC-216). Cinco dosis de inyecciones semanales de ETC-216 administradas a pacientes con SCA mostraron su efecto al conseguir la regresión de la aterosclerosis coronaria (hasta un 4,5%) medida por IVUS80.
Otras terapias sobre apo-A1
La infusión directa de HDL reconstituida (rHDL, apo-A1 combinada con fosfolípidos) ha demostrado mejorar el TRC124 usando objetivos de valoración bioquímicos. Respecto a los objetivos de valoración de imagen, el ensayo clínico ERASE81 evaluó mediante IVUS el efecto de 4 inyecciones semanales de rHDL en 183 pacientes; en comparación con placebo (objetivo primario: cambio en volumen de ateroma), no se vio efecto alguno, pero en comparación con el valor basal, se observó una tendencia hacia la regresión de la placa (aunque a expensas de ascensos moderados de las enzimas hepáticas). En otro estudio con 20 pacientes con claudicación intermitente, se inyectó una única dosis de rHDL y se realizó aterectomía femoral 5-7 días más tarde. En comparación con el grupo control (infusión salina), los pacientes aleatorizados a infusión de rHDL mostraron menor infiltración lipídica, menor infiltración de macrófagos y valores más bajos de marcadores de inflamación125.
Otra estrategia es el uso de moléculas miméticas de apo-A1. Se trata de moléculas pequeñas (18-22 aminoácidos) cuya estructura es similar a la de apo-A1 (243 aminoácidos). La L-5F se administra por vía intravenosa y ha demostrado reducir la progresión de la aterosclerosis en ratones sin alterar el perfil lipídico126. La D-4F es un compuesto que administrado por vía oral consigue la regresión de la aterosclerosis en ratones127. En humanos, de momento tan sólo se ha estudiado cómo mejora las propiedades antiinflamatorias del cHDL128.
Asimismo, se ha comprobado que las inyecciones semanales de HDL autóloga deslipidada (es decir, sólo las apolipoproteínas) producían una reducción del 12% del volumen de la placa evaluada por IVUS en 28 pacientes que habían sufrido un SCA129 frente a un 3% de aumento de la placa en controles, una diferencia prometedora pero que no alcanzó significación estadística dado el escaso tamaño muestral.
Finalmente, los fosfolípidos forman parte de la molécula de cHDL. En ratones elevan el cHDL y reducen la aterosclerosis130. En 16 voluntarios normolipémicos, el fosfatidilinositol, un derivado de la lecitina de soja, aumenta las concentraciones de cHDL en un 13-18%131.
CONCLUSIONES
Los efectos ateroprotectores del cHDL han quedado suficientemente establecidos tanto desde el punto de vista epidemiológico como preclínico. Respecto a los estudios de intervención con diferentes terapias encaminadas a elevar el cHDL, debemos darnos cuenta de que hay algunas que incrementan la concentración del cHDL, mientras que otras mejoran su funcionalidad y aumentan el TRC. El abordaje de la aterosclerosis mediante la doble estrategia de reducir los valores de cLDL y aumentar los de cHDL se debe considerar como el tratamiento más efectivo para la ECV según las evidencias disponibles.
DECLARACIÓN DE CONFLICTO DE INTERESES
Los autores han declarado no tener ningún conflicto de intereses.
ABREVIATURAS
ACV: accidente cerebrovascular.
CETP: proteína de transferencia de ésteres de colesterol.
cHDL: colesterol asociado a las lipoproteínas de alta densidad.
cLDL: colesterol asociado a las lipoproteínas de baja densidad.
ECV: enfermedad cardiovascular.
GIM: grosor íntima-media.
IAM: infarto agudo de miocardio.
ICC: insuficiencia cardiaca.
TRC: transporte reverso de colesterol.
Correspondencia: Dr. J.J. Badimón.
Atherothrombosis Research Unit. The Zena and Michael A. Wiener Cardiovascular Institute.
1 Gustave Levy Place, Box 1030. Mount Sinai School of Medicine. New York, NY 10029, USA.
Correo electrónico: juan.badimon@mssm.edu