
Distintas enfermedades específicas pueden causar miocardiopatía dilatada, aunque en la mayoría de los casos no se consigue identificar causa alguna y se la considera idiopática. La diferenciación entre formas idiopáticas y secundarias es esencial, dado que algunas de estas son potencialmente reversibles.
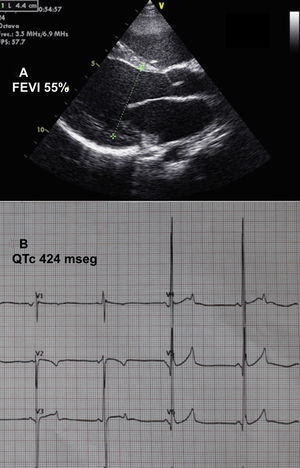
Se presenta el caso de una niña de 11 años, adoptada al año de vida desde Kazajistán. A los 2 años ingresó en cuidados intensivos de otro hospital por un cuadro de dificultad respiratoria, y se diagnosticó miocardiopatía dilatada secundaria a posible miocarditis. Asociaba talla baja (puntuación Z, –3). Se la trató con inhibidores de la enzima de conversión de la angiotensina, diuréticos, digital y bloqueadores beta, y se la derivó a nuestra unidad a los 10 años de edad por empeoramiento de su grado funcional. En la ecocardiografía, el ventrículo izquierdo presentaba dilatación grave, con diámetro telediastólico de 54 mm (puntuación Z,+6) y una fracción de eyección del ventrículo izquierdo del 40% (figura 1A). Destacaba trabeculación miocárdica marcada, sin cumplir criterios de no compactación y regurgitación mitral moderada-grave funcional. La resonancia magnética confirmó los hallazgos ecocardiográficos y demostró retención patológica de gadolinio subepicárdica en la región anterolateral. En el electrocardiograma destacaba bradicardia sinusal grave con ondas T anormales, picudas y con voltajes muy altos en las derivaciones precordiales medias, así como un intervalo QT extremadamente corto (QTc, 265 ms) (figura 1B). Se realizó Holter de 24 h, que no registró ninguna arritmia.
 y electrocardiografía (B) antes del tratamiento. FEVI: fracción de eyección del ventrículo izquierdo.")
Debido a la excepcional asociación del síndrome QT corto (habitualmente enfermedad puramente eléctrica) con miocardiopatía, se estudió una etiología común que explicara la coexistencia de ambas condiciones. Se realizó una búsqueda bibliográfica sobre causas de QT corto, y entre ellas se encontraron el déficit primario de carnitina (DPC) y otras alteraciones de la beta-oxidación de los ácidos grasos. Tras extracción de estudio metabólico, se inició tratamiento empírico con levo-carnitina. El amonio en sangre resultó en 365 μmol/l (valores normales, 10-60μmol/l); la creatincinasa, 260 U/l (valores normales, 0-145), y la carnitina, 1,3 μmol/l (diagnóstico,<5; valores normales, 14-69,7). Se incrementó progresivamente la dosis de carnitina hasta 200 mg/kg/día ajustándola según las concentraciones. En la ecocardiografía, tras 6 meses de tratamiento, el ventrículo izquierdo se redujo, la contractilidad mejoró (figura 2A) y quedó una regurgitación mitral trivial. El intervalo QT se normalizó (figura 2B). La paciente se encuentra asintomática y se han suspendido los fármacos para la insuficiencia cardiaca.
 y electrocardiografía (B) después del tratamiento. FEVI: fracción de eyección del ventrículo izquierdo.")
Se realizó estudio genético por secuenciación masiva (NGS), que detectó la variante c.865C>T (p.Arg289*), un truncamiento patogénico en el gen SLC22A5, en heterocigosis simple. Dicha variante se describió previamente en otro paciente con DPC en combinación con otra variante patogénica1. Se analizó una muestra de fibroblastos y se demostró una clara disminución del transporte de carnitina intracelular respecto a los controles, con lo que se confirmó la sospecha de DPC.
El DPC es una enfermedad genética rara, autosómica recesiva, que afecta a 1:40.000-1:120.000 personas2. Se debe a la presencia de 2 alelos mutados (bien en homocigosis o 2 mutaciones en heterocigosis compuesta) en SLC22A5, que codifica el transportador OCTN2, encargado del transporte de carnitina dentro de las células. La carnitina es un cofactor esencial para que los ácidos grasos de cadena larga pasen a través de la membrana interna de las mitocondrias para la beta-oxidación. Los ácidos grasos no utilizados se acumulan en los tejidos afectados. Esta enfermedad tiene un amplio espectro de manifestaciones clínicas, como miopatía, hepatomegalia, hiperamoniemia, crisis recurrentes de hipoglucemia y miocardiopatía hipertrófica o dilatada, que puede ser su única manifestación2,3. Esta afección se incluye en el cribado neonatal en algunas comunidades autónomas de España.
En nuestro caso, pese a no contar con una segunda variante patogénica, el estudio en fibroblastos confirmó la sospecha de DPC. Se desconoce si pudieran existir alteraciones genéticas poco habituales, no detectables por NGS, que alteren el comportamiento de este gen a nivel proteico (variantes intrónicas profundas, en regiones promotoras o que afecten al splicing), por lo que son necesarios estudios funcionales para confirmar esta hipótesis.
En cuanto a las características del electrocardiograma, hace décadas se describieron alteraciones en el DPC, con ondas T anormales, picudas en derivaciones precordiales medias4 similares a las secundarias a hiperpotasemia. Otros autores han descrito a pacientes con DPC, miocardiopatía y acortamiento del intervalo QT3,5. Roussel et al.5 comprobaron en un modelo animal de ratones que el tratamiento con Mildronate (que induce deficiencia de carnitina) reproducía el fenotipo con aparición de miocardiopatía (hipertrófica) y QT corto. Algunos autores señalan que la predisposición a la muerte súbita de los pacientes con defectos de la beta-oxidación de ácidos grasos, como el DPC, se debe a arritmias por alteración de la repolarización eléctrica secundarias a disfunción de canales iónicos. Ferro et al.6 demostraron que una alta concentración de acilcarnitinas de cadena larga altera la corriente de potasio del Ikr.
El caso que se presenta lleva a 2 conclusiones. En primer lugar, ante todo paciente afectado de miocardiopatía dilatada, es esencial descartar siempre etiologías potencialmente tratables, máxime en niños. En el caso de que sea una enfermedad hereditaria, se podrá asesorar a otros familiares. En segundo lugar, ante todo paciente en el cual concurran QT corto y miocardiopatía dilatada, debe sospecharse un DPC, ya que, de diagnosticarse a tiempo, se puede tratar y revertir completamente la afección cardiaca1,3-5.