
La enfermedad de Fabry (EF) es un trastorno ligado al cromosoma X, debido a mutaciones en el gen GLA, que produce un déficit de la enzima alfagalactosidasa A, encargada de degradar determinados glucoesfingolípidos. El déficit enzimático hace que estos se acumulen, lo que lleva a una disfunción de órganos vitales (fundamentalmente riñón y corazón) y una muerte prematura. Las mujeres pueden presentar el mismo grado de afección que los varones, pero suele ser menos grave y más tardía. Se la considera una enfermedad rara, con una frecuencia de 1:40.000-1:120.0001.
Su diagnóstico es un reto y muchas veces es el cardiólogo, que estudia al paciente por una miocardiopatía hipertrófica (MCH), quien la sospecha. El estudio genético confirma el diagnóstico y permite el estudio familiar; sin embargo, hay variantes genéticas de patogenicidad dudosa1. Este hecho es crucial, ya que hay un tratamiento enzimático sustitutivo (TES) caro y no exento de complicaciones.
Se presenta el caso de una familia con EF en la que se demostró que la variante p.Arg118Cys no causa la enfermedad, por lo que se recalca la necesidad de estar alerta ante variantes similares.
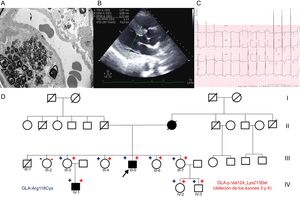
A un varón de 45 años, con proteinuria desde los 36 y cuya función renal se fue deteriorando, se le realizó una biopsia renal. El microscopio electrónico mostró «cuerpos de cebra» sospechosos de EF (figura A). Ante estos hallazgos, se derivó al paciente a la unidad de cardiopatías familiares, donde se completó el estudio, en el que destacaban una grave hipertrofia del ventrículo izquierdo (HVI) en el ecocardiograma y PR corto e HVI en el electrocardiograma (figuras B y C). Se realizó estudio genético, panel de 16 genes relacionados con la MCH (incluido GLA), con next generation sequencing. Se detectó la variante p.Arg118Cys, pero también una deleción de los exones 3 y 4 (p.Val124_Lys213del).

A: microscopia electrónica de biopsia renal con «cuerpos de cebra». B: ecocardiograma del caso índice con HVI. C: electrocardiograma del caso índice con HVI y PR corto. D: árbol genealógico; cuadrado: varón; círculo: mujer; en negro: fenotípicamente afectados; flecha: probando; diagonal: fallecido; + rojo: portador de la deleción de los exones 3 y 4; – rojo: no portador de la deleción de los exones 3 y 4; + azul: portador de la variante p.Arg118Cys; – azul: no portador de la variante p.Arg118Cys; HVI: hipertrofia del ventrículo izquierdo. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
El estudio familiar (figura D) mostró que su madre tenía MCH y falleció de insuficiencia renal a los 64 años. Algunos familiares eran de otra comunidad autónoma, por lo que se contactó con otra unidad de cardiopatías familiares. Se detectó a 5 mujeres y 2 varones portadores, 1 de ellos un niño de 4 años (las características clínicas se resumen en la tabla). Todos los portadores de la deleción en GLA también son portadores de la variante p.Arg118Cys, mientras que los no portadores no lo son. El estudio familiar permitió establecer que tanto la variante de tipo missense como la deleción se encuentran en el mismo alelo (figura A).
Datos clínicos y genéticos de la familia estudiada
| Paciente | Sexo | Portadores p.Arg118Cys | Portadores deleción | Actividad enzima GLA* | Edad al diagnóstico (años) | Afección cardiaca | Afección renal | Córnea verticilata | Angioqueratomas | Crisis de Fabry |
|---|---|---|---|---|---|---|---|---|---|---|
| III:5 | V | + | + | 0,1 | 43 | HVI | IR | + | – | + |
| III:2 | M | – | – | 3,5 | – | – | – | – | – | |
| III:3 | M | + | + | 2,0 | 51 | HVI | Microalbuminuria | + | – | + |
| III:4 | M | + | + | 2,1 | – | – | – | – | – | |
| III:6 | M | + | + | ND | – | – | – | – | – | |
| III:7 | M | + | + | 1 | – | – | – | – | – | |
| IV:1 | V | + | + | ND | 26 | HVI | Microalbuminuria | – | + | + |
| IV:2 | M | + | + | 1,0 | – | – | – | – | – | |
| IV:3 | V | + | + | 0,1 | 4 | – | – | – | – | – |
–: sin afección; +: afección; crisis de Fabry: dolor urente en manos y pies; HVI: hipertrofia del ventrículo izquierdo; IR: insuficiencia renal; M: mujer; ND: no disponible; V: varón.
Ya se habían descrito deleciones similares y en relación con la EF que no aparecen en bases de datos de población normal. La deleción se produce por un reordenamiento entre secuencias repetitivas denominadas Alu, las cuales se distribuyen por todo el gen. Si esta variante se tradujera, se perdería parte del dominio en el que se localiza el sitio de unión al sustrato, con lo que la proteína carecería de actividad enzimática.
Se publicó previamente que la p.Arg118Cys era responsable de la EF2, pero los artículos no confirman la cosegregación familiar y dan solo información de casos índice. Además, son estudios de tipo Sanger, por lo que no se puede descartar que hubiera deleciones causantes de la enfermedad indetectables con este método. Esta limitación se soluciona con la next generation sequencing con plataforma que incluya estudio de copy number variations.
La p.Arg118Cys también aparece en la población normal: en la base de datos Exome Aggregation Consortium, se identifica en 8 varones hemicigotos de una población de control3. El aparecer en bases de datos de genotipado de población general con una frecuencia elevada (superando la prevalencia de la EF en población general) apoya la no patogenicidad de la misma. Ferreira et al.4 demostraron que los portadores de p.Arg118Cys tienen una sobrevida similar a de la población de control y no sufren complicaciones de la EF. Es importante destacar que la actividad enzimática puede estar disminuida, pero probablemente sea suficiente para que la enzima ejerza su función, por lo que no se debe utilizar esta medición para definir la patogenicidad y, menos aún, para iniciar TES.
En la familia estudiada, el caso índice está recibiendo TES, y se ha ofrecido este tratamiento a 1 sobrino (IV:1) y 1 hermana (III:2). El resto de las mujeres portadoras están asintomáticas y sin afección. El niño de 4 años está asintomático, pero en estrecho seguimiento, debido a la posibilidad de iniciar TES precoz para evitar el daño de órganos diana.
En resumen, la EF debe incluirse en el diagnóstico diferencial de la MCH y, si está indicado, realizar un estudio genético mediante next generation sequencing que permita detectar deleciones. Debe ponerse en duda la patogenicidad de las variantes previamente consideradas patogénicas, como la p.Arg118Cys, en estudios en los que las deleciones pudieron pasar inadvertidas y en los que no se confirmó la cosegregación familiar. El estudio integral de la familia y un adecuado estudio genético permiten solventar estas dificultades5.
CONFLICTO DE INTERESESL. Monserrat-Iglesias es accionista de Health in Code S.L.