La secuenciación masiva (o de nueva generación) del ácido desoxirribonucleico ha revolucionado el diagnóstico genético. Esta tecnología reduce el trabajo y el coste necesarios para el análisis simultáneo de muchos genes, lo que hace que más pacientes puedan acceder a un estudio genético. En el caso de la miocardiopatía hipertrófica, se ha pasado de analizar los tres genes principales (MYH7, MYBPC3, TNNT2) a secuenciar más de veinte genes. A pesar de las ventajas que esto representa en términos de información, muchos pacientes presentan variantes de significado incierto (fundamentalmente cambios de aminoácido) que están también en al menos uno de los controles cuyo genoma se ha secuenciado. Esta situación aboca a un «callejón sin salida» en caso de que no se pueda demostrar que esas variantes segregan con la enfermedad en la familia del paciente. En ausencia de evidencia clara de que sean realmente patogénicas, no se podrán emplear para un consejo genético fiable a los familiares del paciente. Finalmente, la secuenciación masiva también permite identificar nuevos genes candidatos pero, una vez más, el problema de las variantes de significado incierto limita el éxito de estos estudios.
Palabras clave
La miocardiopatía hipertrófica (MCH) es la forma primaria de hipertrofia del ventrículo izquierdo1–3. En el caso de la MCH, el desarrollo de la hipertrofia sería consecuencia de una «presión» contra el músculo cardiaco para incrementar su actividad contráctil, a fin de compensar una reducción de esta capacidad por motivos endógenos4. De estos, el más común sería la presencia de una isoforma anormal de alguna de las proteínas contráctiles, pero también pueden estar en el origen de la MCH alteraciones en los mecanismos de producción energética y reguladores de la contracción miocárdica5. Tratándose de una enfermedad esencial, es lógico que tras la MCH se hallen mutaciones en alguno de los genes que codifican proteínas de la contracción miocárdica6. Al ser una causa frecuente de muerte súbita cardiaca de jóvenes adultos, el conocimiento de la base genética de esta enfermedad es de gran interés social7.
Desde el descubrimiento de que muchos casos presentaban mutaciones en MYH7 y MYBPC3, otros genes mutados en un porcentaje menor de pacientes se han incorporado al arsenal diagnóstico7–12. En el aspecto práctico, en los últimos años se ha producido una revolución tecnológica con la introducción de nuevas técnicas de secuenciación del ácido desoxirribonucleico (ADN) (secuenciación masiva o next generation sequencing), que permiten analizar todos estos genes al mismo tiempo y con menor coste13–16. A pesar de la ventaja que representa acceder a más información sobre la variación genética, esto no siempre permite responder de manera definitiva a la cuestión de si el paciente tiene alguna mutación que explique la enfermedad.
El objetivo de esta revisión es aportar una visión de las ventajas y los inconvenientes que la secuenciación masiva ha introducido en el diagnóstico genético de la MCH.
¿QUÉ SABEMOS DE LA GENÉTICA DE LA MIOCARDIOPATÍA HIPERTRÓFICA?Antes de disponer de técnicas de secuenciación masiva, para determinar la presencia de mutaciones había que amplificar cada gen en múltiples fragmentos y leer cada uno por separado mediante el método de Sanger17,18. Esto requiere gran esfuerzo técnico y económico, por lo que la mayoría de los laboratorios se limitaban a secuenciar los genes más frecuentemente mutados (MYH7, MYBPC3 y TNNT2), y en los casos negativos se podía estudiar otros11,12. A partir de estos estudios se puede extraer varias conclusiones:
- 1.
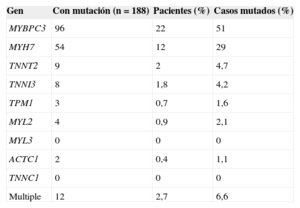
Un 40-60% de los pacientes tendrían mutaciones en alguno de los genes ya asociados a la MCH. Los genes MYH7 y MYBPC3 son los más frecuentemente mutados y representan alrededor del 50% de las mutaciones identificadas (tabla 1).
Tabla 1.Comparación de los resultados del análisis de los principales genes sarcoméricos en 444 pacientes no emparentados estudiados en el Hospital Universitario Central de Asturias
Gen Con mutación (n=188) Pacientes (%) Casos mutados (%) MYBPC3 96 22 51 MYH7 54 12 29 TNNT2 9 2 4,7 TNNI3 8 1,8 4,2 TPM1 3 0,7 1,6 MYL2 4 0,9 2,1 MYL3 0 0 0 ACTC1 2 0,4 1,1 TNNC1 0 0 0 Multiple 12 2,7 6,6 - 2.
Es más probable hallar mutaciones en pacientes con antecedentes familiares y con MCH grave y de aparición precoz. Por lo tanto, en casos esporádicos, formas no graves y de manifestación a edad avanzada, será menos probable encontrar mutaciones. En estos casos esporádicos también es más probable hallar variantes que presentan dificultades para concluir un papel patogénico, entre otros motivos porque no hay familiares afectados con los que confirmar su transmisión con la enfermedad. Estas variantes se clasifican como de significado incierto (VSI) y constituyen un problema para el consejo genético, dado que no se puede establecer de modo concluyente que sean la causa de la enfermedad en el caso índice (figura 1).
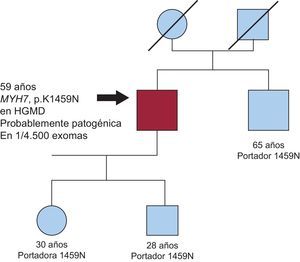
 era un varón diagnosticado a los 69 años con una hipertrofia de 19mm. Tras secuenciar los nueve genes sarcoméricos, resultó ser portador del cambio p.K1459N en MYH7. Se ha hallado en otros pacientes con miocardiopatía hipertrófica y la predicción informática lo clasifica como probablemente patogénico. Está en uno de los exomas secuenciados. Un hermano del paciente era portador de esta variante y permanecía asintomático y sin hipertrofia a la edad de 65 años. Con esta información no se puede concluir que sea una mutación relacionada con la miocardiopatía hipertrófica en lugar de un polimorfismo «raro» (no patogénico). HGMD: Human Gene Mutation Database.") Figura 1.
Figura 1.Familia con portadores de una variante de significado «incierto» en el gen MYH7. El caso índice (flecha) era un varón diagnosticado a los 69 años con una hipertrofia de 19mm. Tras secuenciar los nueve genes sarcoméricos, resultó ser portador del cambio p.K1459N en MYH7. Se ha hallado en otros pacientes con miocardiopatía hipertrófica y la predicción informática lo clasifica como probablemente patogénico. Está en uno de los exomas secuenciados. Un hermano del paciente era portador de esta variante y permanecía asintomático y sin hipertrofia a la edad de 65 años. Con esta información no se puede concluir que sea una mutación relacionada con la miocardiopatía hipertrófica en lugar de un polimorfismo «raro» (no patogénico). HGMD: Human Gene Mutation Database.
(0.09MB). - 3.
En contra de lo que indicaban los primeros estudios, no habría una relación clara entre el gen mutado y la gravedad de la MCH. En general, es mejor hablar del grado de gravedad de una mutación concreta19. Ahora bien, para establecer esta relación entre una mutación (genotipo) y el fenotipo, es necesario un número mínimo de pacientes portadores, lo que solo es posible con unas pocas mutaciones como la deleción de 25 nucleótidos en MYBPC3, p.R453C y p.G716R en MYH7 o p.R92W en TNNT2, todas claramente patogénicas y de mal pronóstico7,20–23. Estas mutaciones están distribuidas por todo el mundo, pero hay otras que son exclusivas de regiones concretas y también puede haber pacientes suficientes para analizar su relación con el fenotipo. Por ejemplo, en Asturias el 8% de los casos (no emparentados) son portadores de un codón de parada en MYBPC3 (p.G263X) asociado a un fenotipo generalmente benigno24. En Islandia, el 58% de los pacientes serían portadores de la mutación MYBPC3 c.927-2A>G, relacionada con un fenotipo heterogéneo25. Una mutación en la primera base del intrón 23 de MYBPC3 es frecuente en los pacientes españoles, y se caracterizaría por una MCH a mediana edad y mal pronóstico con alto riesgo de muerte súbita cardiaca26.
- 4.
Muchas variantes clasificadas como posibles mutaciones son exclusivas de un paciente y su familia (mutaciones privadas). En estos casos, puede ser imposible establecer una relación genotipo-fenotipo, y es uno de los motivos de que las guías clínicas desaconsejen emplearlas para definir el riesgo y tomar decisiones terapéuticas para los portadores3,8,27,28. Un obstáculo para la interpretación de los resultados es que para la MCH no hay actualmente una base de datos que recoja todas las mutaciones conocidas, y solo hay acceso a través de lo publicado o de la suscripción a la base HGMD (Human Gene Mutation Database). Por lo tanto, un laboratorio puede clasificar como nuevas muchas variantes que, en realidad, ya han sido halladas (y no publicadas) en otros pacientes.
- 5.
Hasta un 5% de los pacientes con MCH pueden ser portadores de dos o más mutaciones. En nuestra serie de 444 pacientes, 12 (3%) eran dobles portadores de los genes MYH7, MYBPC3 o TNNT212. Otros grupos han descrito un porcentaje similar (el 2% en la serie de la Clínica Mayo)11. La presencia de dos o más mutaciones en un paciente se ha relacionado con mayor gravedad de la enfermedad29,30. De hecho, las guías clínicas consideran la presencia de más de una mutación como un factor de mal pronóstico, por lo que sería admisible emplear esta información genética para la toma de decisiones terapéuticas3,8.
 era un varón diagnosticado a los 69 años con una hipertrofia de 19mm. Tras secuenciar los nueve genes sarcoméricos, resultó ser portador del cambio p.K1459N en MYH7. Se ha hallado en otros pacientes con miocardiopatía hipertrófica y la predicción informática lo clasifica como probablemente patogénico. Está en uno de los exomas secuenciados. Un hermano del paciente era portador de esta variante y permanecía asintomático y sin hipertrofia a la edad de 65 años. Con esta información no se puede concluir que sea una mutación relacionada con la miocardiopatía hipertrófica en lugar de un polimorfismo «raro» (no patogénico). HGMD: Human Gene Mutation Database.")
Hay situaciones particulares en las que se debe valorar la presencia de más de una mutación en una familia, aun cuando en el caso índice solo se haya encontrado una mutación (figura 2). Cuando haya afectados en la familia que no sean portadores, se puede sospechar que estos podrían tener una segunda mutación. Asimismo, la presencia de dos mutaciones podría explicar la heterogeneidad en la manifestación fenotípica en una misma familia, en la cual los portadores de una sola manifestarían un fenotipo menos grave que los dobles portadores. Para incrementar el rendimiento del estudio genético en una familia, sería aconsejable buscar las mutaciones en el paciente con la MCH más grave y a edad más precoz.
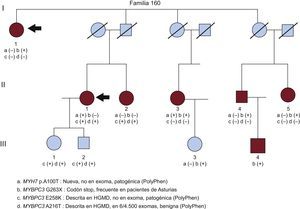
 era una mujer portadora de la mutación p.G263X en MYBPC3 (variante b). Sin embargo, una sobrina afectada (II.1) no era portadora, por lo que se secuenciaron en ella los nueve genes y se hallaron tres variantes, de las que MYH7 p.A100T (a) y MYBPC3 p.E258K (c) serían probablemente patogénicas. HGMD: Human Gene Mutation Database.")
Familia con varios cambios posiblemente patogénicos. El caso índice (I.1) era una mujer portadora de la mutación p.G263X en MYBPC3 (variante b). Sin embargo, una sobrina afectada (II.1) no era portadora, por lo que se secuenciaron en ella los nueve genes y se hallaron tres variantes, de las que MYH7 p.A100T (a) y MYBPC3 p.E258K (c) serían probablemente patogénicas. HGMD: Human Gene Mutation Database.
Un aspecto particularmente intrigante es la posibilidad de que muchos pacientes con formas de MCH aparentemente esporádicas en realidad sean portadores de dos o más mutaciones de penetrancia reducida. Es decir, variantes con efecto patogénico «débil» que por sí solas predisponen a formas de la enfermedad poco graves, pero que combinadas incrementarían su gravedad. Los portadores de una sola mutación podrían permanecer asintomáticos a edad avanzada.
LAS MUTACIONES HABLAN POR SÍ SOLASAl abordar el estudio genético de un paciente con MCH, se puede prever la probabilidad (alta o baja) de encontrar alguna mutación. Es decir, una información clara sobre la causa genética de la enfermedad. Simplemente, se debe tener en cuenta si hay varios afectados con MCH confirmada en la familia o si se trata de un caso esporádico. Si a la historia familiar se añade un fenotipo grave y presentación a edad temprana, hay alta probabilidad de encontrar un cambio que se clasifique como mutación. Son estas familias con muchos afectados las que han permitido identificar los genes sarcoméricos ligados a la MCH31–34.
En los pacientes que no cumplan estas condiciones, es muy probable que no se hallen mutaciones o que se encuentre alguna VSI11,12. No habrá muchas dudas sobre el comportamiento patogénico de un cambio de base en el ADN que se traduzca en un cambio de un aminoácido por un codón de parada, o uno en las primeras o últimas bases de un intrón (mutaciones de ayuste o splicing que afectan al procesamiento del ácido ribonucleico dando un mensajero y una proteína anómala). En la MCH, la mayor parte de los cambios de nucleótido conllevan un cambio de un aminoácido por otro (cambios de sentido o variantes missense). Hay cambios de este tipo que no dejan lugar a dudas sobre su carácter patogénico, como las anteriormente citadas p.R453C y p.G716R en MYH7 o p.R92W en TNNT2. Entre otras razones, porque hay varios pacientes/familias con estos cambios que no han sido hallados en controles sanos. La mayoría de estos cambios se encuentran en familias con varios afectados, y una vez identificados no representa ningún problema el consejo genético sobre los portadores (predispuestos a padecer la enfermedad) y los no portadores (que tendrían el mismo riesgo de sufrir MCH que el resto de la población)35. Incluso se puede plantear un diagnóstico preimplantacional para la descendencia de estos portadores36. Estos cambios en el ADN, mutaciones bona fide, se podría decir que «hablan por sí mismas» por el contexto familiar y fenotípico en el que se encuentran (figura 3).
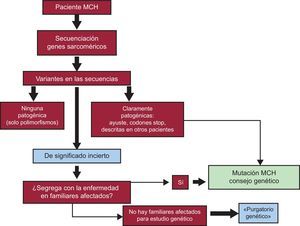
 pueden considerarse la causa de la enfermedad en el paciente y sus familiares portadores. Para las variantes de significado incierto (mayoritariamente cambios de aminoácidos), es necesario demostrar que segregan con la enfermedad, lo que implicaría disponer de varios afectados en la familia. En ausencia de estos afectados, muchas variantes quedarán en una situación de indefinición patológica (el «purgatorio genético»). MCH: miocardiopatía hipertrófica.")
Esquema de los pasos a seguir desde el estudio genético en un paciente al consejo genético en sus familiares. Las variantes claramente patogénicas (por su efecto, cambios intrónicos de splicing o ayuste, introducción de codones de parada prematuros, etc.) pueden considerarse la causa de la enfermedad en el paciente y sus familiares portadores. Para las variantes de significado incierto (mayoritariamente cambios de aminoácidos), es necesario demostrar que segregan con la enfermedad, lo que implicaría disponer de varios afectados en la familia. En ausencia de estos afectados, muchas variantes quedarán en una situación de indefinición patológica (el «purgatorio genético»). MCH: miocardiopatía hipertrófica.
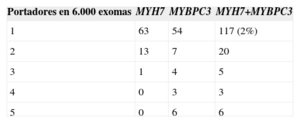
Hasta hace poco, nos hemos desenvuelto en una «edad de la inocencia» genética, en la que cualquier cambio de aminoácido hallado en un paciente con MCH que no estaba en un grupo de controles (usualmente entre 100 y 500) se clasificaba como la mutación causante de la enfermedad en ese caso. A los familiares que eran también portadores se les recomendaba seguimiento y a los no portadores se les reducía el riesgo de afección en el futuro. Si había portadores asintomáticos a edad avanzada, se podía concluir que la mutación tenía una penetrancia reducida, lo que seguramente implicaría un carácter benigno. En algunas familias aparecía algún paciente con MCH que no era portador, lo que indicaba la existencia de una segunda mutación en ese caso. Sin embargo, en los últimos años se ha acumulado información sobre la variación genética en miles de controles, hasta llegar a los más de cinco mil exomas secuenciados (el exoma es la secuencia del genoma que codifica proteínas, alrededor del 5% de los tres mil millones de nucleótidos). Al buscar nuestras mutaciones sarcoméricas en estas bases de datos, nos hemos dado de bruces con la cruda realidad: muchas estaban en al menos uno de esos controles (tabla 2)37. Surgen entonces las dudas: ¿hasta qué grado aquella variante que se consideraba una mutación no sería realmente un polimorfismo «raro» sin relación con la enfermedad?
Número de portadores de variantes nucleotídicas «raras» que darían lugar a cambios de aminoácido, presentes en solo 1-5 exomas secuenciados
| Portadores en 6.000 exomas | MYH7 | MYBPC3 | MYH7+MYBPC3 |
|---|---|---|---|
| 1 | 63 | 54 | 117 (2%) |
| 2 | 13 | 7 | 20 |
| 3 | 1 | 4 | 5 |
| 4 | 0 | 3 | 3 |
| 5 | 0 | 6 | 6 |
El término «purgatorio genético» ha sido acuñado por Michael J. Ackerman (cardiólogo de la Clínica Mayo) para hacer referencia a la situación de indefinición en la que quedarían los cambios de aminoácido que, hallándose en un paciente, están también en alguno de los exomas secuenciados38. Aunque en su ensayo se refiere a los cambios en los genes del síndrome de QT largo, la idea puede extenderse a la MCH y a la mayoría de las enfermedades hereditarias. Estas variantes pasan a ser de «significado patológico incierto» (VSI), es decir, puede que sean la causa de la enfermedad… o puede que no. Y esta incertidumbre conlleva problemas para el consejo genético. ¿Qué valor tiene analizarlas en los familiares del paciente si no se puede concluir su carácter patogénico? Excluir el riesgo de sufrir la enfermedad de alguien no portador tendría consecuencias si en realidad hay otra mutación no identificada (en otro gen) de la que sí fuese portador. Y un portador de la VSI al que se hubiera recomendado seguimiento o incluso tratamiento podría ser no portador de la mutación (que, en realidad, no ha sido identificada).
De un total de seis mil exomas secuenciados, en MYH7 hay 63 cambios de aminoácido únicos y 54 en MYBPC3 (tabla 2). Es decir, hay 119 controles que son portadores de un cambio que solo estaba en ellos. El escenario se vuelve más complejo si se añaden los demás genes sarcoméricos más frecuentemente mutados (tabla 1), que suman 60 variantes únicas. Si todas estas variantes fuesen patogénicas por el simple hecho de su rareza, el 3% de la población tendría una mutación en alguno de los nueve genes principales de la MCH, lo que multiplicaría por 15 la prevalencia estimada de la enfermedad (1/500=0,2%)39. La situación podría ser aún más «caótica» a medida que se añaden genes al análisis, hasta llegar a un punto en el que es casi seguro que se va a encontrar al menos un cambio «sospechoso» en algún gen que, de forma más o menos concluyente, se ha relacionado con la hipertrofia cardiaca.
A favor de que un cambio de aminoácido sea una mutación puede jugar el hecho de que algún algoritmo bioinformático (de los varios que tratan de definir la patogenicidad) lo clasifique como tal. Estos programas emplean varios criterios, y el grado de conservación del aminoácido entre las especies es uno de los que recibe más peso siguiendo el «dogma» de que lo que la evolución conserva debe ser funcionalmente importante, y cualquier cambio a ese nivel probablemente va a tener consecuencias40. Sin embargo, estas no dejan de ser predicciones y una variante clasificada como «probablemente patogénica» puede ser benigna en realidad40. De hecho, si se admitiese como mutaciones todas las variantes raras en los genes sarcoméricos y que el programa Polyphen-2 clasifica como patogénicas, la prevalencia de la MCH debería ser mucho mayor que la estimada41. Un escenario similar se presenta con otras cardiomiopatías, como la dilatada o la arritmogenia del ventrículo derecho.
Hay que recalcar que el «purgatorio genético» es un lugar de transición a la espera de que alguien saque esas variantes de la indefinición en que se hallan38. Seguramente muchas son de verdad patogénicas, pero otras no, y mientras no se pueda concluir su relación con la enfermedad, habrá que hablar al paciente y sus familiares de un «quizá, hacen falta más estudios; mientras tanto mantendremos el seguimiento cardiológico».
¿CÓMO SALIR DEL «PURGATORIO GENÉTICO»?Aun cuando un cambio de aminoácido hallado en un paciente aparezca en las bases de datos del exoma, su carácter patogénico no es descartable. Siempre se puede pensar que el control portador, aparentemente sano, en realidad podría tener hipertrofia del ventrículo izquierdo subclínica. Puede tratarse de un sujeto joven y, si la variante es de penetrancia reducida, aún no se verían sus efectos. Aunque son necesarios datos del exoma de gran número de personas sanas y de edad avanzada, se podría seguir en una situación poco clarificadora. En un análisis de los genes MYH7 y MYBPC3 en 300 sujetos mayores de 70 años y sin manifestación de cardiopatía (reclutados a través de varios centros de salud del Principado de Asturias), se han hallado variantes únicas en 9 (3%). Obviamente, una ecocardiografía podría mostrar hipertrofia del ventrículo izquierdo en los controles sanos portadores de cambios de aminoácido raros, como se ha demostrado en un estudio en las poblaciones de Framingham y Jackson Heart42. Incluso se podría recurrir a métodos más «finos», como la resonancia magnética, para buscar indicios de la enfermedad43.
Los porcentajes de VSI en pacientes con MCH y en la población general indican que muchas de estas variantes raras tienen que ser realmente patogénicas. En nuestra serie de pacientes (n=444), 50 (11%) eran portadores de cambios de aminoácidos hallados solo en ellos y, de estos, 35 no figuraban en la base del exoma. Si un 11% de los pacientes tienen variantes raras frente al 3% de la población general, necesariamente muchas de estas variantes tendrán un efecto patogénico. Pero, como ya se ha señalado, que la variante no aparezca en las bases del exoma no permite concluir su comportamiento patogénico, ya que siempre puede ser un polimorfismo raro presente en ese paciente.
¿Cómo sacar esas VSI del purgatorio genético? Para concluir la relación con la MCH de estas variantes de significado incierto sería necesario demostrar su segregación con la enfermedad en la familia del caso índice o realizar estudios funcionales que demuestren su carácter patogénico.
SegregaciónUna variante patogénica hallada en un paciente debería hallarse en todos los afectados de su familia. Como ya se ha señalado, las mutaciones con un efecto patogénico claro van a dar formas graves de MCH y de aparición precoz, por lo que es muy probable que haya antecedentes familiares y haya disponibles varios afectados para confirmar la segregación. Por otro lado, muchas de estas variantes se han encontrado en varias familias y es poco probable que estén en controles sanos y figuren así en las bases de datos del exoma. Por el contrario, es más probable hallar VSI en pacientes sin antecedentes familiares de MCH, a los que frecuentemente se diagnostica a edad avanzada. Aunque estas variantes sean patogénicas, su penetrancia sería reducida y se podrían hallar en los controles, especialmente si estos tienen una edad por debajo de la de manifestación de los síntomas. Dado que la demostración de la patogenicidad a través de su segregación con la enfermedad sigue una regla estadística, depende del número de afectados que se estudie en una familia y de su grado de parentesco.
BioquímicosUna variante patogénica debería ser «visible» a través de estudios moleculares a escala de la proteína. Por ejemplo, introduciendo el gen mutado en células cultivadas, se debería ver que su proteína se procesa de manera anormal formando agregados y alterando el fenotipo de la célula. Si el tejido cardiaco del paciente está disponible, se tendría que observar el mismo comportamiento en esa proteína.
AnimalesTambién se podría demostrar su efecto patogénico «creando» un animal transgénico (generalmente un ratón) que expresase la proteína mutada y debería desarrollar un fenotipo de MCH44. Esta opción es muy costosa y actualmente inabordable para todas las VSI.
«MÁS MADERA»: BUSCANDO NUEVOS GENES EN LA MIOCARDIOPATÍA HIPERTRÓFICAA pesar de que actualmente hay más de 20 genes que podrían estar mutados en los pacientes con MCH, en un elevado porcentaje de los pacientes no se halla ninguna variante que pudiera explicar la enfermedad9,11,12. Volvemos a recalcar que esta situación es más probable en pacientes aparentemente esporádicos o sin antecedentes familiares claros. Esto implica que deberían existir otros genes relacionados con la MCH, y la cuestión es cómo descubrirlos entre los aproximadamente 25 mil genes del genoma humano. Una tarea hasta hace poco difícil de abordar, incluso limitándose a genes que codifican proteínas que participan en la fisiología cardiaca. Sin embargo, ahora es asequible analizando el exoma, aunque no sea una labor exenta de dificultades a la hora de interpretar los resultados.
En primer lugar, hay que asumir que, al secuenciar el exoma de una persona, se va a identificar centenares de variantes «posiblemente patogénicas» y en varios genes. Incluso limitándose a los de «valor» cardiaco, se identificarán varios genes candidatos. Una vez más, hay que asumir que la patogenicidad de esas variantes se define en gran medida por su ausencia en controles, con las limitaciones que esto representa. En ausencia de una segregación de la «posible mutación» con la enfermedad en la familia del paciente, podría ser imposible demostrar su relación con la MCH.
De lo que se acaba de decir, puede colegirse que, para buscar nuevos genes, además de partir de casos sin mutación en genes ya relacionados con la MCH, se debe disponer de varios afectados en la familia, que deberían ser también portadores de la posible mutación identificada en el caso índice. Cumplirán estos criterios solo unas pocas familias, ya que la mayoría de los casos en que buscar nuevos genes serán esporádicos o no tendrán varios familiares afectados disponibles para su estudio genético. En último término, se podrá reducir el número de mutaciones/genes candidatos a unos pocos, incluso a uno solo si se dispone de muchos afectados en la familia en los que verificar la presencia de la posible mutación. En el caso de la MCH, apenas hay estudios de búsqueda de nuevos genes mediante secuenciación del exoma, probablemente porque, aunque muchos pacientes no presenten mutaciones en los genes ya relacionados con la enfermedad, son muy pocos los casos de familias con varios afectados en los que confirmar las mutaciones.
Una vez seleccionados el gen o los genes candidatos, es necesario realizar estudios funcionales para demostrar que la mutación tiene efecto en la proteína o que esta se halla alterada en el tejido cardiaco de esos pacientes. Siguiendo esta aproximación experimental, nuestro grupo ha identificado a la filamina C como nuevo gen (FLNC) mutado en los pacientes con MCH45.
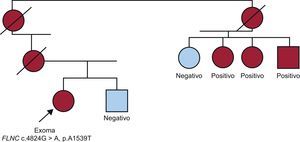
EL CASO DE LA FILAMINA CSe partió de una paciente con MCH y varios afectados en su familia, y se disponía de ADN de tres de ellos (figura 4). La paciente era negativa para mutaciones en los principales genes sarcoméricos12. Tras secuenciar su exoma, se identificaron más de 100 posibles mutaciones en otros tantos genes. Es decir, cambios en la secuencia de su ADN que darían cambios de aminoácido no descritos en los controles de la base del exoma. Se pudo restringir el número de candidatos a 25 tras considerar solo los genes que codificaban proteínas relacionadas con la fisiología cardiaca, y a solo uno tras determinar si esas variantes estaban también en el resto de los familiares afectados: solo el cambio c.C4824G en el gen FLNC, que se traduciría en un cambio de aminoácido alanina > treonina (p.A1539T), era compartido por los cuatro afectados45.

Familia en la que se identificó FLNC como causante de la miocardiopatía hipertrófica. El caso índice era una mujer sin mutaciones en los principales genes sarcoméricos. Tras secuenciar su exoma, se descartó la presencia de mutaciones en todos los genes conocidos ligados a la miocardiopatía hipertrófica. De las posibles mutaciones en genes relacionados con la fisiología cardiaca, solo la de la filamina C estaba en todos los familiares afectados.
La filamina C es una proteína que se expresa principalmente en el músculo estriado, donde interaccionaría con los filamentos de actina y podría funcionar comunicando la membrana celular con el propio sarcómero46. Para demostrar que el cambio de aminoácido tendría consecuencias en la función de la proteína, se creó por mutagénesis dirigida una copia del gen que contenía el cambio, y se la introdujo en miocitos cardiacos de rata, que luego se cultivaron y se sometieron a análisis funcionales. En paralelo se realizaron los mismos análisis con células transfectadas con una copia de FLNC de secuencia normal. Así se pudo demostrar que FNLC con la posible mutación formaba agregados, mientras que la forma normal permanecía en la fracción celular soluble45. Además, se observaron agregados de otras proteínas del citoesqueleto, como la actina. Aunque tanto los datos de la segregación familiar con la MCH como los de los estudios celulares apuntaban claramente a FLNC como causa de la enfermedad, un hallazgo puntual en una sola familia podría tener un valor muy limitado, por lo que lo ideal era demostrar la presencia de posibles mutaciones en otros pacientes con MCH. Por ello, se secuenció el gen FLNC en 92 pacientes sin mutaciones en los principales genes implicados en la MCH, y se halló una posible mutación en 8 casos, de los que 6 tenían al menos un segundo familiar también afectado, y todos ellos eran portadores de la posible mutación.
Más aún, dos de los pacientes se habían sometido a un trasplante, por lo que se disponía de tejido cardiaco para estudio. En ambos casos, el análisis histológico mostró agregados intracelulares de filamina C y sarcómeros con estructura anormal, con desorganización miofibrilar y fibrosis45.
La principal conclusión del estudio es que se podría encontrar posibles mutaciones en FLNC en alrededor del 10% de los pacientes con MCH que no presenten mutaciones en los principales genes ya relacionados con esta enfermedad. Se podría preguntar qué grado de confianza tenemos en que estos cambios de verdad sean mutaciones: la misma que para los demás genes claramente relacionados con la MCH, siempre basada en la ausencia de estas variantes en controles y su segregación con la enfermedad en la familia. Por supuesto, algunas de las posibles mutaciones en FLNC serán cambios no patogénicos, pero si se compara la frecuencia en nuestros pacientes (9/93; 9,7%) con la de variantes únicas y «posiblemente patogénicas» en la base del exoma (91/6.500; 1,4%), la estadística también juega a favor de que este sea un gen implicado en la MCH. El porcentaje de pacientes con posibles mutaciones en FLNC queda sin definir hasta que se publiquen los resultados de series de pacientes más numerosas y de otras poblaciones.
Uno de los mayores problemas que se afrontan al valorar FLNC como candidato en la MCH es que ya se ha relacionado con formas familiares de miopatía fibrilar (afección del músculo esquelético), y algunos pacientes presentan cardiomiopatía además de la característica neuropatía46–48. Ninguno de nuestros pacientes con posibles mutaciones en FLNC tenía miopatía, y en varios se pudo descartar anomalías histológicas en el tejido muscular obtenido mediante biopsia45. Este fenómeno, en el que las mutaciones en un gen pueden dar manifestaciones clínicas heterogéneas, no es excepcional. Como ejemplos, el propio gen MYH7 se ha relacionado con una forma rara de miopatía distal de inicio precoz49; las mutaciones en FHL1 causan generalmente una enfermedad neuromuscular, y a su vez se han asociado con MCH con disfunción diastólica del ventrículo izquierdo sin afección neuromuscular50; las mutaciones en BAG3 se han relacionado con neuropatía de inicio precoz en varias familias, pero también con miocardiopatía dilatada sin afección neuromuscular en otras51,52. Al igual que para estos genes/enfermedades, todo apunta a que las mutaciones en dominios diferentes de la filamina C podrían dar lugar a manifestaciones clínicas de miopatía o a MCH. Sin duda, esta es una línea que requiere investigación básica para conocer el mecanismo molecular por el que FLNC se relaciona con la MCH.
CONCLUSIONESLa secuenciación masiva del ADN ha facilitado la identificación de las bases genéticas de las enfermedades hereditarias haciéndola accesible a un número de pacientes en aumento. En el caso de la MCH, tras hallar una mutación bona fide en un paciente, se puede emplear para el consejo genético en sus familiares, identificando a los portadores que requerirán seguimiento, diagnosticando a nuevos pacientes asintomáticos e incluso derivando en técnicas de diagnóstico preimplantacional. La secuenciación masiva también ha revelado la presencia de muchas variantes «raras» en la población general (controles sin la enfermedad), hasta un grado de complejidad genética que podría dificultar la interpretación de los resultados en muchos pacientes. Estos casos son portadores de variantes de significado incierto, la mayoría de ellas cambios de aminoácido no hallados en otros pacientes. En ausencia de familiares afectados en los que demostrar su segregación con la enfermedad, estas variantes se clasifican como «de significado incierto», ya que podrían ser no patogénicas (polimorfismos raros). Sin resolver este dilema no se pueden emplear de manera segura para el consejo genético de la familia del paciente.
La secuenciación masiva también permite buscar mutaciones en genes nuevos analizando el genoma de pacientes en los que no se haya encontrado mutaciones en los genes ya relacionados con la MCH. La existencia de las variantes de significado incierto obliga a buscar esos genes nuevos en casos con varios familiares afectados, en los que se debe confirmar la presencia de las posibles mutaciones.
CONFLICTO DE INTERESESNinguno.