Muchas enfermedades neuromusculares afectan al corazón, generalmente de manera asintomática, pero en ocasiones con relevancia clínica. En este grupo se incluirían las distrofias musculares, las miopatías congénitas, inflamatorias y metabólicas, algunas enfermedades heredodegenerativas, enfermedades de la unión neuromuscular y neuropatías periféricas adquiridas o hereditarias1. Un correcto diagnóstico requiere de un equipo multidisciplinario experimentado con un alto grado de sospecha clínica y que dirija la realización de un estudio del músculo esquelético y el nervio periférico con enzimas musculares, estudio neurofisiológico y, si es necesario, biopsia muscular. Se recomienda una evaluación exhaustiva de la función respiratoria, especialmente si los pacientes presentan alteraciones clínicas, ya que puede determinar su pronóstico o ser su clínica predominante. Las anomalías cardiacas más prevalentes son las miocardiopatías, las arritmias ventriculares y los trastornos del sistema de conducción con asociación a muerte súbita cardiaca2. Sin embargo, la evidencia disponible es escasa y con series de pacientes aisladas, con recomendaciones de actuación basadas en opiniones de expertos3.
Se pretende describir las manifestaciones cardiacas y la penetrancia familiar en nuestra serie de pacientes diagnosticados genéticamente.
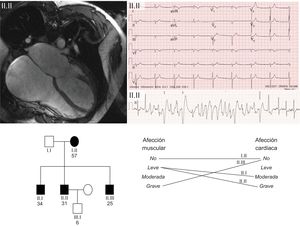
Se incluyó a un total de 11 pacientes pertenecientes a 7 familias, con una media de edad de 36,18 ± 13,4 años y predominio de varones (72%). La tabla muestra un resumen clínico y genético de los 11 casos. La expresión clínica más frecuente fue la miocardiopatía dilatada con disfunción sistólica del ventrículo izquierdo. Hubo una elevada prevalencia de trastornos del ritmo (81,8%), con mayor frecuencia entre las distrofias de Steinert y Emery-Dreifuss. El 100% de los pacientes con enfermedad de Steinert presentaron trastornos del ritmo y el 75% de los que tenían Emery-Dreifuss2. Un caso presentaba un antecedente familiar de muerte súbita. La figura muestra un ejemplo de familia con distrofia de Emery-Dreifuss.
Características clínicas y genéticas de los 11 pacientes
| Sexo | Edad (años) | N.o de familia | Pedigrí del sujeto | Familiares estudiados, n | Familiares afectados, n | Mutación | Enfermedad muscular | Ecocardiografía | Expresión muscular | ECG | CK (U/l) | Estudio neurofisiológico | Biopsia | Resonancia magnética | Tratamiento médico | Marcapasos/TRC/DAI | Tamaño auricular |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Varón | 31 | 1 | Sujeto II.II | 3 | 2 | Emerina (Gln219) | Emery-Dreifuss | MCD FEVI ligeramente deprimida | Ligera | Ritmo nodular. QRS ancho | 1.116 | Patrón miopático | Cambios miopáticos crónicos distróficos por probable denervación miógena | Realce tadío mesocárdico septal | Ramipril 5 mg/24 h, bisoprolol 5 mg/24 h | DAI-TRC | Aurícula aneurismática. Parálisis auricular |
| Varón | 57 | 1 | Sujeto I.II | 3 | 2 | Emerina (Gln219) | Emery-Dreifuss | Normal | No | Ritmo sinusal. QRS estrecho | 156 | Sin signos patológicos | No disponible | No disponible | No | No | No dilatada |
| Varón | 34 | 1 | Sujeto II.I | 3 | 2 | Emerina (Gln219) | Emery-Dreifuss | MCD FEVI normal | Ligera | Ritmo nodular. QRS estrecho | 766 | Patrón miopático | No disponible | Realce tardío mesocárdico en región septal basal y media | Ramipril 2,5 mg/24 h, bisoprolol 2,5 mg/24 h | No | Aurícula aneurismática. Parálisis auricular |
| Varón | 25 | 1 | Sujeto II.III | 3 | 2 | Emerina (Gln219) | Emery-Dreifuss | Normal | Moderada | Ritmo sinusal. QRS estrecho | 180 | Patrón miopático | No disponible | No disponible | No | No | No dilatada |
| Varón | 34 | 2 | Sujeto II.I | 2 | 1 | Negativa | Atrofia muscular espinal | MCD FEVI normal | Grave | Ritmo sinusal. QRS estrecho | 452 | Patrón neuropático con signos de denervación | Atrofia con componente neurógeno | Realce tardío mesocárdico septal y subepicárdico en VI | No | No | No dilatada |
| Varón | 30 | 2 | Sujeto II.II | 2 | 1 | Negativa | Atrofia muscular espinal | MCD FEVI ligeramente deprimida | No | Ritmo sinusal. QRS estrecho | 116 | Patrón neuropático | No disponible | Realce tardío subepicárdico en todos los segmentos del VI | Ramipril 2,5 mg/24 h, bisoprolol 2,5 mg/24 h | No | No dilatada |
| Varón | 28 | 3 | Sujeto I.I | 0 | 0 | Negativa | Duchenne | MCD FEVI gravemente deprimida | Grave | Ritmo sinusal. QRS estrecho | 612 | Patrón miopático | Fenotipo Duchenne/Becker, predominante Duchenne | No disponible | Enalapril 2,5 mg/24 h, carvedilol 6,25 mg/12 h, eplerenona 25 mg/24 h | DAI | Ligeramente dilatada |
| Varón | 38 | 4 | Sujeto I.I | 0 | 0 | DMD (Lys2400) | Becker | MCD FEVI gravemente deprimida | Moderada | Ritmo sinusal. QRS estrecho | 505 | Patrón mixto. Aumento de actividad. Fibrilaciones | Necrosis celular en múltiples segmentos. Déficit parcial de distrofina | Realce tardío mesoepicárdico y subepicádico extenso y difuso biventricular | Ramipril 2,5 mg/24 h, bisoprolol 10 mg/24 h, eplerenona 25 mg/24 h | DAI | Moderadamente dilatada |
| Mujer | 20 | 5 | Sujeto II.I | 3 | 1 | DMD (Xp21) | Duchenne | MCD FEVI gravemente deprimida | Grave | Ritmo sinusal. QRS estrecho | 773 | Patrón miopático | No disponible | Realce tardío subepicárdico parcheado en VI | Ramipril 10 mg/24 h, bisoprolol 7,5 mg/24 h, eplerenona 25 mg/24 h | DAI-TRC | Moderadamente dilatada |
| Mujer | 65 | 6 | Sujeto I.II | 2 | 1 | DMPK (CTG) | Steinert | FEVI moderadamente deprimida. Diámetros normales | Ligera | Flutter auricular común. QRS ancho | 387 | Patrón miopático | No disponible | No disponible | Ramipril 10 mg/24 h, bisoprolol 10 mg/24 h, eplerenona 25 mg/24 h | No | Moderadamente dilatada |
| Mujer | 36 | 7 | Sujeto II.II | 5 | 3 | DMPK (CTG) | Steinert | Normal | Moderada | Ritmo sinusal. QRS ancho | 188 | Patrón miopático | No disponible | No disponible | No | Marcapasos | No dilatada |
CK: creatincinasa; DAI: desfibrilador automático implantable; ECG: electrocardiograma; FEVI: fracción de eyección del ventrículo izquierdo; MCD: miocardiopatía dilatada; TRC: terapia de resincronización cardiaca; VI: ventrículo izquierdo.

Familia con distrofia de Emery-Dreifuss con mutación novel en la emerina. En la imagen superior izquierda se aprecia resonancia magnética cardiaca del caso índice con dilatación biauricular grave. Arriba a la derecha su electrocardiograma basal con ritmo nodular debido a «parálisis auricular» y desarrollo de taquicardia polimórfica en el electrocardiograma de esfuerzo. En la imagen inferior se muestra su pedigrí: nótese la escasa correlación presente entre el grado de gravedad de la afección cardiaca y del músculo esquelético entre los 4 portadores de la mutación. Los cuadrados corresponden a varones y los círculos, a mujeres. En negro, los sujetos enfermos y en blanco, los sanos.
La mayoría de los pacientes (81,8%) sufrían daño muscular de algún grado. El 27,3% manifestaba afección ligera e igual porcentaje, moderada o grave. En nuestra muestra no se halló relación estadísticamente significativa entre el daño muscular catalogado como leve, moderado o grave (en función de los síntomas, signos clínicos y la exploración neuromuscular con estudio electromiográfico realizado por neurología) y la afección cardiaca catalogada por la fracción de eyección en ligera, moderada o gravemente deprimida (correlación de Spearman, ρ = 0,301; p > 0,05). Tampoco hubo relación significativa entre las concentraciones de creatincinasa (CPK) y la afección cardiaca (ρ = 0,54; p > 0,05) la CPK y el daño muscular (ρ = 0,43; p > 0,05).
Respecto a la afección respiratoria, el 18% de nuestros pacientes necesitaron seguimiento respiratorio especializado (1 caso precisó ventilación mecánica invasiva).
El 54,5% de los pacientes tenían tratamiento médico para la insuficiencia cardiaca con bloqueadores beta e inhibidores de la enzima de conversión de la angiotensina, y se añadían antialdosterónicos o diuréticos de asa si era necesario.
El 36% de los pacientes eran portadores de desfibrilador automático implantable, de los cuales el 50% tenía resincronización. El 9% son portadores de marcapasos sin desfibrilador ni resincronización.
Desde la perspectiva familiar, a raíz de los casos índice, se estudió a 15 familiares, de los que 8 estaban afectados; 3 probandos no presentaban mutación causal mediante test genético.
Hay escasas recomendaciones con bajo nivel de evidencia en el tratamiento de estos pacientes3. Por ejemplo, en la estratificación del riesgo arrítmico para implantar un desfibrilador automático implantable o marcapasos. En Steinert, distrofia de cinturas 1B o Emery-Dreifuss, se recomienda implantar un desfibrilador automático implantable cuando haya arritmias ventriculares o indicación de estimulación con nivel de evidencia y grado de recomendación II B4. Además, en la distrofia de cinturas, Steinert o Kearns-Sayre, se considera implantar marcapasos permanente ante cualquier grado de bloqueo auriculoventricular, incluso de primer grado. Nuestros datos concuerdan con esto, pues muestran una elevada prevalencia de trastornos del ritmo. Otro aspecto controvertido es la anticoagulación en pacientes con parálisis auricular (típico de Emery-Dreifuss) o en presencia de arritmias supraventriculares. En una de nuestras familias, esta situación ocurre en 2 varones afectados, con volúmenes auriculares aumentados y sin actividad eléctrica auricular en el electrocardiograma (ECG) ni en registros endocavitarios. Esta situación podría predisponer a mayor riesgo de accidente cerebrovascular, pero no hay evidencia que permita establecer una recomendación sobre la pertinencia de la anticoagulación oral para estos pacientes.
En nuestra muestra no se obtuvieron valores estadísticamente significativos, probablemente por el reducido tamaño muestral, aunque se observó una baja correlación entre la gravedad del daño muscular y la afección cardiaca. Sin embargo, nuestros resultados están influidos porque, en trastornos como la distrofia de Emery-Dreifuss, la afección cardiaca puede preceder a la muscular. La disponibilidad del test genético permite un estudio familiar en cascada e identificar casos con afección cardiaca potencialmente graves que podrían beneficiarse de un tratamiento precoz aunque no muestren daño evidente de músculo esquelético.
Los pacientes con manifestaciones cardiacas más graves son derivados con más frecuencia a nuestra unidad, lo que supone un sesgo de selección. Por ello se precisa cautela al establecer conclusiones generales de prevalencia o modo de afección cardiaca.
Los pacientes diagnosticados de daño neuromuscular deben pasar por cribado cardiovascular mediante historia clínica y familiar, exploración, ECG y ecocardiograma. A aquellos sin afección cardiaca, se les recomienda seguimiento periódico con ECG y ecocardiografía3 a intervalos ajustados a las peculiaridades de cada entidad en concreto. Cardiología debe revaluar al paciente cuando aparezcan nuevos síntomas o alteraciones no conocidas en las pruebas acometidas. También se debe cribar a los familiares de primer grado en el momento del diagnóstico3.
En conclusión, la complejidad y la escasa prevalencia de las enfermedades neuromusculares requieren un mayor control clínico y actuación de equipos multidisciplinarios experimentados, entre los que se debería incluir a un cardiólogo.