
La distrofia miotónica tipo 1 se caracteriza por afección muscular y manifestaciones sistémicas, entre ellas las cardiacas. Nuestro objetivo es documentar la frecuencia y la gravedad de la afección cardiovascular (aparición de disfunción ventricular izquierda y trastornos del ritmo o conducción), la necesidad de implantar marcapasos o desfibrilador o de realizar estudio electrofisiológico y la aparición de muerte súbita durante el seguimiento.
MétodosEstudio observacional retrospectivo de los pacientes con distrofia miotónica tipo 1 remitidos a una consulta monográfica de cardiología y sometidos a seguimiento clínico, electrocardiográfico (con registro Holter) y ecocardiográfico.
ResultadosSe incluyó a 81 pacientes (el 51,9% varones; media de edad, 29,9 ± 14,8 años). El seguimiento medio fue de 5,7 ± 3,9 (1-20) años y se documentó bradicardia sinusal en el 48,8%, disfunción sinusal en el 13,8%, arritmias supraventriculares en el 10%, intervalo PR ≥ 220 ms en el 31,3%, taquicardia ventricular el 5%, intervalo QT corregido largo en el 5%, bloqueo auriculoventricular de segundo o tercer grado en el 8,8% e intervalo QRS ≥ 120 ms en el 7,5%. Sólo 1 paciente presentó disfunción ventricular grave. Durante el seguimiento se implantaron 15 marcapasos y 2 desfibribladores y se realizaron 5 estudios electrofisiológicos, la mayoría por taquicardia ventricular. Sólo se produjo 1 muerte súbita.
ConclusionesLos trastornos de conducción y del ritmo son frecuentes durante la evolución de dichos pacientes, y un porcentaje considerable requiere estudio electrofisiológico e implante de dispositivos (marcapasos o desfibrilador). La disfunción sistólica y la muerte súbita son excepcionales en nuestra experiencia.
Palabras clave
La distrofia miotónica tipo 1 o enfermedad de Steinert es actualmente la distrofia muscular más frecuente en adultos. Es una enfermedad multisistémica hereditaria autosómica dominante con una expresión fenotípica muy variable debida a una repetición inestable del trinucleótido CTG en el brazo largo del cromosoma 19 (19q.21.3), que codifica una proteincinasa. Se asocia a miotonía, debilidad muscular progresiva con atrofia de la musculatura esquelética y numerosas manifestaciones sistémicas, de las que las más frecuentes son aparición de cataratas, diabetes mellitus, hipogammaglobulinemia, retraso mental y afección cardiaca1. La causa más frecuente de mortalidad se debe a insuficiencia respiratoria (hasta un 43%), seguida de eventos cardiacos (hasta un 20%)2.
La afección cardiaca se debe a fibrosis miocárdica y degeneración del sistema de conducción, que además crea un sustrato para arritmias por reentrada y contribuye a la disfunción ventricular que pueden presentar estos enfermos. Por lo tanto, las manifestaciones cardiovasculares se caracterizan fundamentalmente por alteración de la función ventricular izquierda (tanto sistólica como diastólica, aunque gran parte de estos pacientes no refieran clínica de insuficiencia cardiaca)3 y, más frecuentemente, alteraciones electrocardiográficas (hasta el 30,2% de los pacientes)4,5: prolongación de los intervalos PR y QT, ensanchamiento del intervalo QRS, aparición de disfunción sinusal, bloqueos auriculoventriculares (BAV) de segundo y tercer grado, bloqueo de rama derecha o izquierda y taquiarritmias auriculares o ventriculares6.
Varios estudios asocian las anomalías basales en el electrocardiograma con el riesgo de muerte súbita6, lo que lleva al implante de marcapasos (MCP) en un 4,1-11% y de desfibrilador automático implantable (DAI) en un 1,1-5,3% de los casos7, sobre todo cuando se considera el estudio Holter8,9. Actualmente se estima el riesgo de muerte súbita, la necesidad de implantar MCP y DAI por año de seguimiento en el 0,56, el 1 y el 0,2% respectivamente8.
Ante la frecuencia de los trastornos del ritmo o de la conducción desarrollados en la evolución y el riesgo de muerte súbita que conlleva y dado que todavía no hay consenso establecido para el seguimiento de estos pacientes8,9, algunos autores recomiendan la realización de técnicas diagnósticas invasivas ante el hallazgo de alteraciones en el electrocardiograma basal aunque estén asintomáticos, con la finalidad de aumentar la supervivencia4,10,11. El objetivo de nuestro trabajo es documentar la prevalencia y la gravedad de la afección cardiovascular, analizar la necesidad de implantar MCP o DAI o de realizar estudios electrofisiológicos (EEF) y la aparición de muerte súbita durante el seguimiento de los pacientes.
MÉTODOSRealizamos un estudio observacional retrospectivo de los pacientes con diagnóstico de distrofia miotónica tipo 1 remitidos entre 1983 y 2010 desde Neurología a una consulta monográfica de Cardiología del Hospital Universitario Nuestra Señora de Candelaria, centro de tercer nivel de la isla de Tenerife, con una población de referencia de 424.923 habitantes (veinte municipios de la zona sur de Tenerife y la población de las islas de La Gomera y El Hierro). El diagnóstico definitivo de enfermedad de Steinert se realizó mediante estudio genético ante una clínica neurológica sospechosa (mediante reacción en cadena de la polimerasa o Southern blot cuando el fenotipo era manifiesto).
En la consulta de Cardiología realizamos en la primera visita historia clínica cardiológica, electrocardiograma basal, estudio Holter y ecocardiografía. El seguimiento posterior consistió en historia clínica y electrocardiograma anual, y cada 2 años nuevo estudio Holter y ecocardiografía, salvo que la clínica, los trastornos electrocardiográficos o los hallazgos ecocardiográficos indicaran realizarlo antes.
Analizamos las manifestaciones sistémicas de la enfermedad, los antecedentes familiares de muerte súbita y el grado de afección muscular (Muscular Impairment Rating Scale, de Mathieu et al12) en cinco grados: 1, no hay debilidad muscular; 2, mínimos signos sin debilidad distal salvo en los músculos flexores distales de las extremidades inferiores; 3, debilidad distal sin afección proximal salvo para los extensores de los codos; 4, debilidad proximal moderada, y 5, gran debilidad proximal.
Determinamos la fracción de eyección del ventrículo izquierdo mediante ecocardiografía utilizando el método de Teichholz y se consideró que había disfunción ventricular en caso de fracción de eyección del ventrículo izquierdo < 50%.
En el electrocardiograma basal realizado en la consulta, medimos los siguientes parámetros electrocardiográficos: frecuencia cardiaca, intervalo PR, anchura del intervalo QRS e intervalo QT corregido. Definimos bradicardia sinusal como ritmo sinusal < 60 lpm o pausas sinusales diurnas > 3 s, e intervalo QT corregido largo al > 450 ms en varones y > 470 ms en mujeres. Revisamos todos los estudios Holter en búsqueda de taquiarritmias supraventriculares (principalmente fibrilación o aleteo auricular), BAV II o III, disfunción sinusal (presencia de bradicardia sinusal y pausas diurnas > 3 s asociadas o no a taquicardia supraventricular) y taquicardias ventriculares (TV) no sostenidas o sostenidas.
Asimismo, documentamos la necesidad de implantación de MCP o DAI y la realización de EEF. Finalmente, analizamos la mortalidad de los pacientes durante el seguimiento.
Análisis estadísticoUtilizamos la prueba de Kolmogorov-Smirnov para determinar distribución normal de las variables continuas. Las variables categóricas se representan en n (%); las numéricas, como media ± desviación estándar en caso de distribución normal o como mediana (intervalo) en caso contrario. Calculamos la probabilidad de supervivencia libre de MCP mediante el método de Kaplan-Meier. El software empleado para el análisis es el programa SPSS, versión 15.0.
RESULTADOSEn el periodo de estudio se remitió a nuestra consulta a 81 pacientes con diagnóstico genético de distrofia miotónica tipo 1. El 51,9% eran varones, con una media de edad al diagnóstico de la enfermedad de 29,9 ± 14,8 años. El 86,4% tenía antecedentes familiares de la enfermedad y el 16%, antecedentes familiares de muerte súbita. La afección neurológica que se objetivó con mayor frecuencia fue la de grado 2 de la Muscular Impairment Rating Scale (38,7%), y entre las manifestaciones extracardiacas destacó la aparición de cataratas (49,4%) e hipogammaglobulinemia (45,3%). El 78,8% requirió valoración por el Servicio de Neumología para estudiar la necesidad de ventilación mecánica no invasiva. En la tabla 1 se muestran las características epidemiológicas y clínicas de la serie.
Características demográficas y manifestaciones extracardiacas y clínicas del grupo de pacientes remitidos con diagnostico genético de distrofia miotónica tipo 1
| Pacientes, n | 81 |
| Edad (años) | 29,9 ± 14,8 |
| Varones | 51,9 |
| Antecedentes familiares de enfermedad de Steinert | 86,4 |
| Antecedentes de muerte súbita | 16 |
| Presentación congénita de la enfermedad | 4,9 |
| Retraso mental | 23,5 |
| Grado de afección muscular (escala de Mathieu et al12) | |
| 1 | 14,7 |
| 2 | 38,7 |
| 3 | 17,3 |
| 4 | 24 |
| 5 | 5,3 |
| Hipogammaglobulinemia | 45,3 |
| Cataratas | 49,4 |
| Alteraciones gastrointestinales | 27,2 |
| Diabetes mellitus | 16 |
| Necesidad de evaluación neumológica | 78,8 |
| Clínica cardiaca referida por el enfermo | |
| Asintomáticos | 64,6 |
| Mareos | 21,5 |
| Síncope | 6,3 |
| Palpitaciones | 7,6 |
Los datos expresan n (%) o media ± desviación estándar.
Durante un seguimiento medio de 5,7 ± 3,9 (1-20) años el 64,6% se mantuvo sin síntomas cardiovasculares; el 21,5% refirió mareos; el 6,3%, síncope y el 7,6%, palpitaciones. Ningún paciente presentó clínica compatible con insuficiencia cardiaca y sólo 1 paciente (1,3%) mostró disfunción ventricular grave. No objetivamos deterioro de la fracción de eyección del ventrículo izquierdo en los demás pacientes.
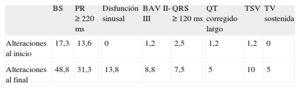
En la primera visita, el 71,6% de los pacientes tenía un registro electrocardiográfico basal normal y se documentó bradicardia sinusal en el 17,3%, intervalo PR ≥ 220 ms en el 13,6%, BAV de segundo grado Mobitz I en el 1,2%, intervalo QRS ≥ 120 ms en el 2,5% e intervalo QT corregido largo en el 1,2%. En 1 paciente (1,2%) objetivamos taquicardia supraventricular (aleteo auricular típico con frecuencia ventricular normal). Sin embargo, al final del seguimiento sólo registramos a 29 pacientes (36,7%) sin alteraciones del ritmo o de la conducción, mientras que presentaron bradicardia sinusal el 48,8%, intervalo PR ≥ 220 ms el 31,3%, intervalo QRS ≥ 120 ms el 7,5% e intervalo QT corregido largo el 5%. Asimismo, durante la evolución 11 pacientes presentaron disfunción sinusal; 8, taquicardia supraventricular; 4, TV sostenida y 7, BAV II-III. Señalar que sólo 7 pacientes (8,9%) del grupo presentaron alguna anomalía desde el inicio que no se modificó a lo largo de la evolución, y el electrocardiograma en el seguimiento tenía las mismas características que el inicial. La tabla 2 muestra las alteraciones electrocardiográficas evolutivas de la serie.
Alteraciones electrocardiográficas objetivadas durante el seguimiento en el grupo estudiado (%)
| BS | PR ≥ 220 ms | Disfunción sinusal | BAV II-III | QRS ≥ 120 ms | QT corregido largo | TSV | TV sostenida | |
| Alteraciones al inicio | 17,3 | 13,6 | 0 | 1,2 | 2,5 | 1,2 | 1,2 | 0 |
| Alteraciones al final | 48,8 | 31,3 | 13,8 | 8,8 | 7,5 | 5 | 10 | 5 |
BAV: bloqueo auriculoventricular; BS: bradicardia sinusal; TSV: taquiarritmias supraventriculares; TV: taquicardia ventricular.
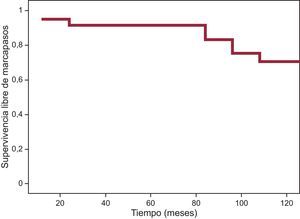
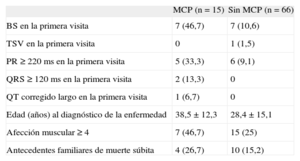
Durante el periodo estudiado, se implantó MCP a 15 pacientes (20%), bicameral en el 93,3% de los casos. Dicho grupo presentaba con mayor frecuencia antecedentes familiares de muerte súbita, afección muscular avanzada y alteraciones electrocardiográficas basales desde la primera visita. La tabla 3 muestra las características de este grupo. Sólo 1 paciente estaba clínicamente asintomático, mientras que el 73,3% refirió mareos, 2 (13,3%) sufrieron síncope y 1, palpitaciones. La indicación del implante se realizó ante los hallazgos electrocardiográficos: para 11 pacientes el motivo fue disfunción sinusal; en 3, BAV II-III, y en 1, se implantó por bloqueo alternante de rama (se realizó EEF, que mostró intervalo HV patológico). El registro Holter resultó una herramienta diagnóstica determinante en 9 casos (documentó en 3 pacientes BAV II-III, bloqueo alternante de rama derecha y de rama izquierda en 1 paciente y disfunción sinusal en el resto). La probabilidad estimada de supervivencia libre de MCP fue del 83,4 ± 5,5% a 90 meses (figura).
Características electrocardiográficas y clínicas de los pacientes que requirieron implante de marcapasos durante la evolución
| MCP (n = 15) | Sin MCP (n = 66) | |
| BS en la primera visita | 7 (46,7) | 7 (10,6) |
| TSV en la primera visita | 0 | 1 (1,5) |
| PR ≥ 220 ms en la primera visita | 5 (33,3) | 6 (9,1) |
| QRS ≥ 120 ms en la primera visita | 2 (13,3) | 0 |
| QT corregido largo en la primera visita | 1 (6,7) | 0 |
| Edad (años) al diagnóstico de la enfermedad | 38,5 ± 12,3 | 28,4 ± 15,1 |
| Afección muscular ≥ 4 | 7 (46,7) | 15 (25) |
| Antecedentes familiares de muerte súbita | 4 (26,7) | 10 (15,2) |
BS: bradicardia sinusal; MCP: marcapasos; TSV: taquiarritmias supraventriculares.
Los datos expresan n (%) o media ± desviación estándar.

Durante el seguimiento, 2 pacientes (2,7%) requirieron DAI: uno de ellos presentó síncope y disfunción ventricular grave y en el otro, a pesar de estar asintomático, el registro Holter mostró múltiples salvas de TV monomorfa no sostenida; se realizó EEF, que indujo varias TV con ablación de una de ellas, por lo que se decidió el implante del dispositivo.
Realizamos EEF a 5 pacientes (6,8%): en 3, con ablación de rama derecha por TV rama-rama; en 1, ablación de TV, y en 1, se requirió ablación del istmo cavotricuspídeo por aleteo auricular típico. La indicación de dicha prueba fue fundamentalmente ante la clínica referida (2 pacientes sufrieron síncope y otro tuvo mareos, con Holter que mostró bloqueo alternante de rama; en los tres casos se indujo TV en el EEF) y en 2 pacientes, a pesar de estar asintomáticos, por los hallazgos electrocardiográficos (TV monomorfa no sostenida en el registro Holter con posterior inducción de TV en uno y aleteo auricular típico en el otro).
Durante el seguimiento fallecieron 8 pacientes (11%): 4 por insuficiencia respiratoria, uno por carcinoma colorrectal no subsidiario de tratamiento quirúrgico por gran debilidad muscular, uno por traumatismo craneoencefálico secundario a una caída y otro por sepsis cutánea de extremidades inferiores. Sólo 1 paciente sufrió muerte súbita.
DISCUSIÓNEl presente trabajo describe la evolución cardiológica de un grupo de pacientes con enfermedad de Steinert seguidos en una consulta monográfica de cardiología de un hospital de tercer nivel. Estos enfermos presentaron trastornos del ritmo y de la conducción como principal manifestación cardiovascular frente al potencial desarrollo de miocardiopatía.
Durante un seguimiento medio de 5,7 años, la mayoría de los pacientes sufrieron alteraciones electrocardiográficas. Si bien muchas de ellas sólo traducen el deterioro del sistema de conducción inherente a la enfermedad, en un 20% de los casos se consideró necesario implantar MCP. Esta frecuencia es superior a la descrita en otras series: Breton et al13 describen una tasa de implantación de MCP del 3,3% en una cohorte de 428 pacientes, con una mediana de edad de 33 (2-81) años, seguidos durante una media de 11,7 años, y Lindqvist et al14, del 2,7% en 36 pacientes con una media de edad de 45 ± 10 años durante un seguimiento medio de 3 años. La elevada proporción de pacientes a los que se implantó MCP de nuestra serie probablemente se deba al seguimiento regular con estudio Holter, herramienta que resultó determinante para decidir una actitud terapéutica en el 60% de los pacientes. En el trabajo de Cudia et al9, todos los pacientes (n = 245; media de edad, 45 años) fueron seguidos con registro Holter, y se documentó una frecuencia de implante de MCP del 11%.
Dado el potencial riesgo de muerte súbita de estos pacientes6, algunos autores proponen la realización de estudios invasivos en búsqueda de hallazgos que puedan modificar la actitud terapéutica, para evitar la temida aparición de muerte súbita4,10,11,15. Así, en la revisión de McNally et al4 se recomienda la realización de EEF ante alteraciones electrocardiográficas basales (intervalo PR > 240 ms e intervalo QRS > 120 ms), y se considera el registro Holter sólo para los pacientes asintomáticos con electrocardiograma basal normal. En nuestro grupo, los pacientes que requirieron MCP tenían una edad al diagnóstico de la enfermedad menor que la referida en los trabajos reseñados9,14 y con mayor frecuencia alteraciones electrocardiográficas desde la primera visita. Probablemente estos hallazgos traducen un daño intrínseco del sistema de conducción que anteceden a la aparición de disfunción sinusal (alteración de la conducción que llevó al implante de MCP más frecuentemente objetivada) o BAV avanzado. Realizamos EEF a 5 pacientes, justificado principalmente por TV, y las principales causas de mortalidad estuvieron relacionadas con el deterioro muscular. De hecho, sólo uno de ellos murió súbitamente. La baja frecuencia de muerte súbita durante el seguimiento contrasta con lo objetivado en el estudio de Groh et al6, donde se recoge una frecuencia del 33,3% en un grupo de 406 adultos seguidos durante 5,5 años. En dicho trabajo, la realización de un electrocardiograma anual no fue requisito indispensable para el seguimiento, y la necesidad de una valoración cardiológica fue a criterio del médico general. Suponemos que en nuestro trabajo la elevada frecuencia de implantación de MCP tuvo un papel fundamental al prevenir la aparición de bradicardia extrema o asistolia, con lo que se obtuvo un porcentaje menor de muerte súbita. La alta frecuencia de uso de dispositivos está influida por los resultados de la monitorización no invasiva en muchos de ellos, al establecerse desde el inicio del seguimiento una monitorización protocolizada.
Sin embargo, la baja incidencia de disfunción ventricular sistólica en nuestro estudio difiere de otros trabajos. En la revisión de Petri et al8, donde se analizan las manifestaciones cardiovasculares (miocardiopatía y alteraciones del ritmo o de la conducción) de 18 estudios publicados entre 1980 y 2010, se documenta una prevalencia de miocardiopatía primaria del 7,2%. Probablemente la mera determinación de la fracción de eyección del ventrículo izquierdo por ecocardiografía subestime la verdadera incidencia de disfunción ventricular. En este sentido, Ozyigit et al3, tras analizar el flujo Doppler transmitral y transtricuspídeo (estándar y mediante Doppler tisular) de 21 pacientes afectos de distrofia miotónica tipo 1 frente a 21 controles, documentaron que el aumento del índice de Tei (índice de rendimiento miocárdico, que permite evaluar de forma no invasiva las alteraciones de la función sistólica y diastólica) es un método más sensible que el cálculo de la fracción de eyección del ventrículo izquierdo para detectar disfunción ventricular en estos enfermos. De hecho, Lindqvist et al14 analizan por ecocardiografía Doppler y electrocardiograma a 36 pacientes afectos frente a 16 controles, y concluyen que en el grupo afecto las medidas convencionales para el cálculo de la fracción de eyección del ventrículo izquierdo tienen poco valor frente a los tiempos de contracción y relajación isovolumétrica del ventrículo izquierdo estando dichos parámetros alargados. Si bien dichos valores pueden explicarse por las alteraciones electrocardiográficas presentes (intervalo PR e intervalo QRS alargado), en un porcentaje de los casos es una causa de asincronía miocárdica en lugar de eléctrica.
CONCLUSIONESLa mayoría de los pacientes con enfermedad de Steinert tienen alteraciones del ritmo y de la conducción durante la evolución de la enfermedad; en nuestra serie, un 20% requirió MCP y el 2,7%, DAI. Una monitorización electrocardiográfica protocolizada y estrecha es fundamental, ya que en un número no despreciable de casos lleva a una actitud terapéutica determinante. En contraste con las alteraciones del ritmo y la conducción, la disfunción ventricular sistólica objetivada por ecocardiografía es muy infrecuente.
CONFLICTO DE INTERESESNinguno.