



Las manifestaciones clínicas de la aterosclerosis, tales como los síndromes coronarios agudos, los eventos cerebrovasculares y la enfermedad arterial periférica, son las principales causas de morbimortalidad en el mundo. La activación y la agregación plaquetarias son, en última instancia, la causa de la progresión y las presentaciones clínicas de la enfermedad. Por ello, los antiagregantes plaquetarios son un pilar fundamental del tratamiento farmacológico de estos pacientes. Una amplia variedad de receptores de superficie tipo integrinas, familia rica en leucina, receptores acoplados a proteínas G y receptores de tirosincinasa, así como moléculas intraplaquetarias, desencadenan y regulan el proceso de activación/agregación plaquetaria. Todas estas moléculas son dianas potenciales de fármacos antiplaquetarios destinados a prevenir y tratar la trombosis arterial. A pesar del beneficio clínico obtenido con el ácido acetilsalicílico (inhibidor de la ciclooxigenasa), el clopidogrel (antagonista del receptor del ADP P2Y12) y los antagonistas de la glucoproteína IIb/IIIa (abciximab, eptifibatida, tirofibán, lamifibán) al disminuir significativamente el riesgo de episodios aterotrombóticos, la morbimortalidad residual sigue elevada. Por ello los esfuerzos se centran en la búsqueda de nuevos tratamientos antiplaquetarios a fin de mejorar su efectividad y su seguridad. De hecho, nuevos fármacos están en fase de desarrollo y varios han llegado ya a uso clínico. Entre ellos están los nuevos inhibidores de los receptores P2Y12 (prasugrel, ticagrelor, cangrelor y elinogrel), antagonistas del receptor PAR1 de la trombina (vorapaxar, atopaxar) y moléculas de señalización intraplaquetaria. Esta revisión profundiza en los mecanismos de acción del arsenal antiplaquetario actualmente en uso y las nuevas aproximaciones terapéuticas.
Palabras clave
Los antiinflamatorios no esteroideos (AINE), incluido el ácido acetilsalicílico (AAS) (fig. 1), son inhibidores de la enzima ciclooxigenasa 1 (COX-1) y, por lo tanto, inhiben la síntesis del tromboxano A2 (TXA2) (fig. 2). Sin embargo, mientras que el AAS consigue una inactivación por acetilación casi completa (≥ 97%) y persistente (≥ 24h) de la COX plaquetaria (isoforma COX-1), los demás AINE actúan como inhibidores reversibles de esta enzima. Es más, como las plaquetas son células anucleadas y, por lo tanto, incapaces de llevar a cabo la síntesis proteica, no pueden reponer la actividad enzimática, por lo que la inhibición plaquetaria se prolonga durante toda la vida de la plaqueta (7–9 días).


Principales vías de activación y agregación plaquetarias y las consiguientes dianas antiplaquetarias. AA: ácido araquidónico; AC: adenilato ciclasa; ADP: adenosindifosfato; AMP: adenosinmonofosfato; ATP: adenosintrifosfato; COX: ciclooxigenasa; DAG: diacilglicerol; FvW: factor de von Willebrand; GMP: guanidil monofosfato; IP3: 1,4,5-trifosfato; PAR: receptor activado por proteasas; PDE: fosfodiesterasas; PG: prostaglandina; PI3K: fosfatidil-inositol trifosfato; PIP2: fosfoinositol 2; PKA: proteincinasa A; PLA: fosfolipasa A2; PLC: fosfolipasa C; TP: receptor del tromboxano; TXA2: tromboxano A2.
El AAS inhibe la COX al unirse al residuo de arginina-120 (el mismo punto de unión de los AINE) y acetilar una serina clave para la acción catalítica de la enzima (serina 529 para la COX-1 plaquetaria y serina 516 para la COX-2 endotelial) reduciendo la síntesis plaquetaria de TXA2. Otro efecto del AAS en las plaquetas es que disminuye la secreción de gránulos densos implicada en la liberación de sustancias proagregantes y vasoactivas durante la activación plaquetaria. Además, un metabolito de la aspirina, el ácido salicílico, tiene cierto efecto fibrinolítico debido a su interacción con los neutrófilos y monocitos con liberación de enzimas proteolíticas (catepsina G y elastasa). El AAS, además, tiene diversos efectos no plaquetarios, como inhibición de las prostaglandinas, inhibición de la síntesis de interleucina (IL) 6 en los leucocitos y reducción de la actividad de los inhibidores de la óxido nítrico sintasa endotelial (eNOS). Todo ello, se cree, contribuye a explicar por qué sus efectos beneficiosos son mayores de lo que cabría esperar de la simple inhibición plaquetaria dependiente de un agonista relativamente débil como es el TXA2.
El AAS bloquea también la agregación secundaria inducida por la trombina, colágeno o ADP debido a que inhibe la producción plaquetaria de diacilglicerol, aunque este efecto es menos duradero que la acción sobre la COX y es apreciable a dosis muy altas de AAS (> 650mg/día). A estas dosis, el AAS puede producir importantes efectos adversos al inhibir la COX endotelial (isoforma COX-2) y reducirse así la síntesis de PGI2, un importante cardioprotector. Cabe destacar los resultados negativos obtenidos tras la inhibición selectiva de la COX-21. Por ello, las dosis recomendadas de AAS no son capaces de prevenir la agregación plaquetaria desencadenada por la activación de otras vías independientes de la síntesis de TXA2. Esto contribuye a explicar los fracasos terapéuticos observados en ensayos clínicos y en la práctica diaria en relación con el uso de AAS como profilaxis antitrombótica.
A pesar de que inicialmente es recomendable una dosis de AAS de 160mg para bloquear completamente la COX-1 plaquetaria, cantidades menores son capaces de inhibir en casi su totalidad la síntesis de TXA2. Por ello, la dosis de mantenimiento puede reducirse a 81–100mg/día de por vida.
En cuanto al papel de la aspirina en prevención primaria y secundaria, un reciente metaanálisis concluyó que, mientras la administración de aspirina a dosis de 100–150mg/día sí previene la presentación de enfermedades isquémicas (cardiovasculares, cerebrovasculares y arteriales periféricas) en pacientes de alto riesgo (prevención secundaria)2, no ocurre lo mismo en prevención primaria. De hecho, el beneficio del AAS se anula por el riesgo de sufrir algún evento adverso importante por el fármaco, entre los cuales figuran las hemorragias. Así, en prevención primaria, mientras el riesgo del primer infarto se reduce en un 18%, el riesgo de hemorragias extracraneales aumenta en un 54%.
Por el contrario, en prevención secundaria, el AAS se ha convertido en el antiplaquetario de referencia tras la aparición de un evento agudo y su administración debe continuarse indefinidamente, salvo que esté contraindicado por alergia, complicaciones gastrointestinales o hemorragia4. Pese a todo, la aspirina sólo reduce los eventos clínicos en un 30% (aproximadamente). Es más, la aparición cada vez más frecuente de la «resistencia a la aspirina» o el también llamado «fracaso en el tratamiento con aspirina» obliga a profundizar en una mejor comprensión de sus efectos, así como a desarrollar nuevas alternativas terapéuticas. De hecho, estudios basados en la incapacidad del AAS para proteger contra la aparición de complicaciones trombóticas, causar una prolongación del tiempo de sangría, inhibir la agregación plaquetaria ex vivo o inhibir la producción plaquetaria de TXA2 han llegado a establecer que un porcentaje elevado (según estudios, hasta un poco creíble 45%) de los pacientes presentan un «fracaso en el tratamiento», más común en ancianos y mujeres5. Los mecanismos implicados en esta resistencia probablemente sean multifactoriales y podrían clasificarse en factores derivados de fallo en la reducción de la síntesis de TXA2 y factores derivados de fallo en el tratamiento. La insuficiente supresión del TXA2 puede derivar de un aumento en la renovación de plaquetas (transfusiones, cirugía de revascularización coronaria), de una mayor síntesis de TXA2 de fuentes no plaquetarias (células endoteliales, monocitos/macrófagos), de la presencia de interacciones farmacológicas con otros AINE8, así como la presencia de polimorfismos genéticos (COX, TXA2 sintasa). Por otro lado, la activación plaquetaria por vías alternativas a COX-1, la reducción en la absorción, el incremento del metabolismo y la poca adherencia al tratamiento pueden contribuir a explicar el posible fallo del tratamiento.
Hasta la fecha se han realizado pequeños estudios que establecen que la supresión incompleta en la síntesis del TXA2 en presencia de suficiente dosis (resistencia a la aspirina) es un marcador potencial de riesgo cardiovascular. Sin embargo, a pesar de que empiezan a conocerse las implicaciones clínicas asociadas a la resistencia a la aspirina, quedan aún importantes cuestiones por resolver, tales como cuál es la metodología de diagnóstico ideal para la identificación de los pacientes con la condición de resistentes y conocer los factores genéticos y mecanismos celulares que conducen a su presentación. Del mismo modo, son necesarios estudios clínicos que aborden la pauta terapéutica a seguir en pacientes que manifiestan la condición de resistencia, a fin de disminuir el riesgo de eventos adversos. En este sentido, algunos estudios apuntan a que un aumento en la dosis de aspirina no aporta beneficio clínico alguno, pero sí puede conducir a mayor número de complicaciones hemorrágicas7.
TriflusalEl triflusal (trifluorosalicílico) (fig. 1) está relacionado estructuralmente con el ácido acetilsalicílico; sin embargo, a diferencia de este, inhibe la fosfodiesterasa del AMPc y del GMPc, hecho que limita la movilización de calcio y la agregación plaquetaria dependiente de calcio (fig. 2). Además, a diferencia del AAS, tiene una actividad selectiva contra el metabolismo del ácido araquidónico en las plaquetas (inhibiendo la COX-1) (fig. 2) sin apenas afectar a la actividad contra el mismo metabolismo en el endotelio vascular (relativa falta de inhibición de la COX-2)8.
El triflusal se administra por vía oral, se absorbe en el intestino delgado, se une a las proteínas plasmáticas casi en su totalidad (99%) y su biodisponibilidad oscila entre el 83 y el 100%. Durante el paso por el hígado se desacetila y se forma su principal metabolito, el 2-OH-4-trifluorometil ácido benzoico (HTB), que es farmacológicamente activo. La vida media del triflusal en personas sanas es de 0,5 ± 0,1h, mientras que la de HTB es 34,3 ± 5,3h, y su eliminación es principalmente renal.
El triflusal es más potente en la inhibición de COX-1 y en la reducción de TXA2 que HTB, pero es menos potente que el AAS. Sin embargo, HTB potencia el efecto del triflusal en la inhibición de la COX-1, mientras que el metabolito del AAS, el ácido salicílico, compite con el compuesto de origen por su unión a la enzima. Por otra parte, el triflusal, a través de la estimulación de la forma constitutiva de la enzima NO sintasa, aumenta la producción de NO por los neutrófilos en un 150%, mientras que AAS lo hace en un 60%. Gracias a esta multiplicidad de acciones, el triflusal presenta actividad antiplaquetaria comparable a la del AAS, pero con un perfil de seguridad más favorable. De hecho, a pesar de tener muchos menos ensayos clínicos que avalen su eficacia, ha demostrado similares beneficios clínicos que el AAS en pacientes con infarto agudo de miocardio9 o que habían sufrido eventos cerebrovasculares isquémicos, con menor incidencia de hemorragia.
Inhibidores dell tromboxanoUna posible limitación en la eficacia antiplaquetaria del AAS está relacionada con la existencia de fuentes no plaquetarias capaces de sintetizar TXA2 y la presencia de agonistas del receptor del tromboxano que se producen no enzimáticamente a través de un proceso de peroxidación de lípidos catalizada por radicales de oxígeno (p. ej., F2-isoprostanos y prostaglandinas [PG] D, D2, E2, F2a y H2). Estas limitaciones quedan cubiertas con el uso de bloqueadores del receptor del tromboxano o TP.
Los mecanismos mediante los cuales los TP inhiben la activación/ agregación plaquetaria implican la señalización del calcio (fig. 2). Por un lado, los receptores del TP (TPα y TPβ) estimulan la familia de las proteínas Gq, lo que a su vez, tras activar la fosfolipasa C, resulta en la acumulación de inositol-1,4,5-trifosfato (IP3) y diacilglicerol (DAG) y estos, a su vez, favorecen la liberación del calcio desde el retículo endoplásmico y la activación de la proteincinasa C (PKC), respectivamente (fig. 2). El aumento en las concentraciones citoplásmicas de calcio induce la activación de vías de señalización implicadas en la agregación plaquetaria. Por otro lado, los TP estimulan la activación de las proteínas de la familia G12 (G12 y G13), que activan, vía IP3 y DAG, la vía de señalización de Rho implicada en cambios conformacionales de la plaqueta (activación plaquetaria) (fig. 2).
Hallazgos recientes indican que la inhibición directa de TP no sólo tiene efectos antiplaquetarios, sino que también previene el desarrollo de la enfermedad aterosclerótica al impedir la interacción leucocito- endotelio y la vasoconstricción. Nuestro grupo ha demostrado la rápida y eficaz actividad del terutrobán (fig. 1)12,13, un potente inhibidor reversible del TP (fig. 2), en inhibir la trombosis inducida por stent sin la concomitante aparición de un mayor riesgo de sangrado. Es más, se ha demostrado que el terutrobán previene la aterogénesis, causa regresión de la placa aterosclerótica y mejora la función endotelial de pacientes con enfermedad arterial coronaria, propiedades relativamente independientes de los efectos derivados del TXA2 sobre las plaquetas15,16. Sin embargo, un reciente estudio que comparaba la eficacia del terutrobán frente al AAS en 18.000 pacientes con accidente cerebrovascular se suspendió antes de tiempo debido a que no se había observado ninguna prueba de beneficios del terutrobán al compararlo con el AAS.
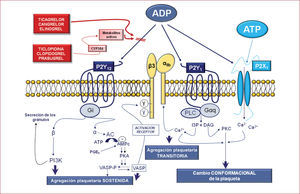
Antagonistas del receptor del adpTiclopidina y clopidogrelMúltiples evidencias experimentales indican que, de todos los agonistas liberados tras la activación plaquetaria, el ADP es uno de los más importantes para reclutar plaquetas y propagar el trombo arterial18. Las plaquetas presentan en su superficie tres receptores para el ADP, P2Y1, P2Y12 y P2X, cada uno de los cuales induce distintas vías de señalización plaquetaria y distintas funciones en la agregación plaquetaria, pero el P2Y12 es la causa última de la agregación plaquetaria persistente (figs. 2 y 3)18. Por todo ello, durante los últimos años los esfuerzos se han centrado en la investigación y desarrollo de fármacos capaces de bloquear este receptor plaquetario19. De hecho, nuevos fármacos están en fase de desarrollo y varios han llegado ya a uso clínico. Entre ellos están los nuevos inhibidores de los receptores P2Y12 prasugrel, ticagrelor, cangrelor y elinogrel (tabla).
Características de los fármacos antagonistas del receptor ADP P2Y12
| Clase fármaco | Conversión metabólica | Reversible | Administración | Vida media | Duración de acción | |
| Clopidogrel | Tienopiridina | Sí | No | Oral | 7h | 5–10días |
| Prasugrel | Tienopiridina | Sí | No | Oral | 3,5h | 5–10días |
| Ticagrelor | Ciclopentil triazopirimidina | No | Sí | Oral | 12h | 1día |
| Cangrelor | Análogo de ATP | No | Sí | Intravenosa | 2-5min | 1h |
| Elinogrel | Antagonista directo de P2Y12 | No | Sí | Oral | 12-14h | 1día |
| Intravenosa | 50min | 2h |
La ticlopidina y el clopidogrel (fig. 1) son agentes antiplaquetarios derivados de las tienopiridinas que antagonizan la agregación plaquetaria inducida por el ADP, y son una eficaz alternativa al AAS en caso de contraindicaciones (alergias, hemorragias). Ambos son profármacos y por ello es necesaria la metabolización por el hígado (fig. 4 y tabla) para convertirse en fármacos activos con propiedades antiplaquetarias. El metabolito activo se une de manera covalente al residuo cisteína de uno de los receptores del ADP (P2Y12), lo que conduce a una modificación irreversible del receptor durante toda la vida de la plaqueta. En el caso del clopidogrel, también se ha demostrado que reduce la formación de conjugados plaqueta-leucocitos en pacientes con SCA21 y la expresión de marcadores inflamatorios en plaquetas activadas como el CD40 ligando y la P-selectina en pacientes sometidos a intervencionismo coronario percutáneo (ICP)22.
La ticlopidina es el primer agente antiplaquetario de la familia de las tienopiridinas que se usó. Dos importantes ensayos clínicos23,24 demostraron la eficacia de la ticlopidina en la reducción de eventos trombóticos en pacientes con enfermedad aterosclerótica. Sin embargo, presentaba un comienzo tardío en su actividad antiplaquetaria y dosis de mantenimiento de 250mg dos veces al día sólo producían una reducción del 50% en la agregación plaquetaria a los 5 días, y un 60–70% a los 8 días, lo cual limitaba su uso en pacientes con síndrome coronario agudo. Además, su uso habitual se vio significativamente mermado por la incidencia, aunque poco frecuente, de neutropenia (8‰). Por ello emergió el clopidogrel como nuevo agente antiplaquetario similar a la ticlopidina, pero con menos efectos adversos (la neutropenia aguda se presentaba con una incidencia del 0,5‰).
El estudio CAPRIE fue el estudio de referencia que confirmó la eficacia antiplaquetaria del clopidogrel con respecto a la aspirina. Este estudio demostró que, en pacientes con historia de infarto de miocardio, infarto cerebral o enfermedad arterial periférica, el clopidogrel reducía la recurrencia de eventos isquémicos en un 8,7% respecto al AAS25. Es más, pasadas 2h tras una administración de carga de 600mg, se observaba una reducción significativa de la agregación plaquetaria, que era máxima pasadas las 6h (40–60% de reducción). En casos de urgencia, sin embargo, ese tiempo necesario para alcanzar una inhibición plaquetaria óptima puede aumentar el riesgo de trombosis aguda. Se planteó la hipótesis de que unas dosis de carga mayores (900mg) acelerarían la actividad antitrombótica del fármaco y se solventaría esa limitación. Sin embargo, se demostró que las dosis de carga de 900mg no se asociaban a mayor velocidad de actuación del fármaco, pero sí a mayor tendencia a hemorragias.
Otra importante limitación del clopidogrel es la gran variabilidad interindividual detectada en cuanto a su capacidad antiagregante, la llamada «resistencia al clopidogrel»28. Incluso se ha identificado un número no despreciable de pacientes que son «no respondedores» al clopidogrel, que presentan un incremento en la incidencia de trombosis subaguda tras la implantación de un stent. El concepto de «variabilidad de respuesta al clopidogrel» ha derivado en la preocupación de que algunos pacientes pueden no estar adecuadamente protegidos de la activación y la agregación plaquetarias y, por lo tanto, estarían expuestos a mayor riesgo de sufrir un evento trombótico30. Hay una heterogeneidad considerable en la actividad de las isoenzimas del citocromo hepático P450 en las poblaciones humanas, y se ha hecho evidente que una minoría considerable de personas (hasta un 14% en algunas series) tiene polimorfismos en CYP2C19 y en CYP3A4 (fig. 4), que causan un deterioro del metabolismo del clopidogrel. Además, la absorción del clopidogrel está regulada por la glucoproteína-P, una proteína dependiente de energía (ATP) que actúa como una bomba extrusora de fármacos a través de las membranas y se localiza, entre otros tejidos, en las células epiteliales del intestino. Por ello, un aumento en su expresión o su función puede derivar en variaciones en la biodisponibilidad de estos fármacos. Todo ello se traduce en niveles bajos de inhibición de las plaquetas en ciertos individuos (los llamados «no respondedores») y resulta en un aumento al triple en el riesgo de eventos adversos cardiovasculares mayores. Sin embargo, los factores que determinan la función plaquetaria y la respuesta al clopidogrel son más complejos que la respuesta debida a un solo polimorfismo, sobre todo en el paciente individual, y si bien muchos centros realizan habitualmente pruebas genéticas para identificar a estos potenciales «no respondedores», este enfoque personalizado no se ha traducido en mejores resultados hasta la fecha, aunque quedan estudios en curso.
Otra limitación asociada al uso de clopidrogrel, especialmente en pacientes que requieren intervenciones quirúrgicas urgentes, radica en su capacidad de inhibir de manera irreversible (tabla) el receptor P2Y12 al formar un puente disulfuro con este receptor, y para revertir sus efectos antiplaquetarios es necesaria la transfusión de plaquetas. Por ello, se recomienda suspender su tratamiento al menos 5 días antes de someterlo a cirugía, tiempo necesario para la suficiente renovación plaquetaria.
PrasugrelEl prasugrel (CS-747, LY640315) es una tienopiridina y por ello es un profármaco. Sin embargo, a diferencia del clopidogrel, la metabolización de prasugrel es más eficiente, requiere un solo paso metabólico de las isoenzimas hepáticas CYP (tras la acción de las esterasas plasmáticas) para convertirse en un agente activo que antagonice de manera irreversible el receptor plaquetario P2Y12 (fig. 4)32,33. Por ello, en comparación con dosis estándar de clopidogrel, produce una inhibición plaquetaria más rápida, pronunciada con dosis inferiores y menos variabilidad de respuesta, por lo que su uso es ventajoso en situaciones que requieren una rápida inhibición plaquetaria. Igualmente, la respuesta al prasugrel no se ve tan afectada por el uso concomitante de inhibidores de CYP. El prasugrel tiene una semivida de 3,5h (tabla).
TicagrelorEl ticagrelor (AZD6140) no pertenece a la familia de las tienopiridinas y es una ciclopentil-triazolopirimidina (CPTP) (fig. 3). Es el primer inhibidor oral reversible que actúa directamente sobre el receptor P2Y12 (fig. 1). A diferencia del clopidogrel y el prasugrel, no es un profármaco, por lo que no requiere metabolización previa y el nivel de inhibición refleja la concentración plasmática del compuesto (fig. 4). Se absorbe rápidamente, alcanza concentraciones plasmáticas máximas 1,5h tras su administración y la desaparición de la acción es rápida, puesto que tiene una semivida de 12h (tabla). Estas diferentes propiedades farmacodinámicas y farmacocinéticas proporcionan al ticagrelor posibles ventajas ante las tienopiridinas, como un efecto antiplaquetario más rápido y potente y con menos variabilidad que el clopidogrel. Otra ventaja es que su excreción es mayoritariamente a través de la bilis y las heces, por lo que no hay necesidad de reducir las dosis en pacientes con insuficiencia renal.


El cangrelor es un potente antagonista reversible del receptor P2Y12 análogo del ATP (fig. 3), con una semivida < 10min (tabla). No es activo cuando se administra por vía oral y por eso, a diferencia de todos los anteriores antagonistas del receptor del ADP, se administra por vía endovenosa y puede desempeñar un papel importante en pacientes en quienes los tratamientos enterales sean difíciles de administrar (pacientes intubados o con hemesis intratables) o requieran una rápida inhibición plaquetaria. De hecho, alcanza un alto grado de inhibición plaquetaria (> 90%) a los pocos minutos tras su administración. Gracias a su mecanismo de acción reversible y de desaparición rápida (una semivida extremadamente breve, 2–5min, a causa de una rápida desactivación por ectonucleotidasas plasmáticas), se observa una recuperación de la función plaquetaria 1–2h tras suspender la infusión del fármaco. El cangrelor ha demostrado en pacientes con SCA una capacidad de inhibición plaquetaria casi completa y en un tiempo comparable al alcanzado por el abciximab (inhibidor de la GPIIb/IIIa). Es más, comparado con el abciximab, se ha demostrado mayor rapidez en el retorno de la función plaquetaria tras la interrupción del tratamiento, lo que le confiere un mejor perfil de seguridad.
ElinogrelEl elinogrel es un antagonista reversible, potente y competitivo del receptor P2Y12 (fig. 3) en fase experimental que está disponible en formulaciones tanto vía endovenosa como vía oral (tabla). No requiere metabolización, presenta una semivida de 12–14h y su excreción es un 50% fecal y un 50% renal.
Antagonistas de la glucoproteína IIb/IIIaLa activación plaquetaria se produce en respuesta a diversos agonistas que actúan por vías metabólicas independientes, pero que convergen en un efector final común: la activación de la integrina GPIIb/IIIa (αIIbβ3, CD41/CD61). Los receptores IIb/IIIa plaquetarios activados son capaces de reconocer y unirse a la secuencia de aminoácidos arginina- glicina-aspartato (RGD) y a la secuencia Lys-Gln-Ala-Gly-Asp-Val (KQAGDV), ambas contenidas en el fibrinógeno (fig. 2). La secuencia RGD se halla también en otras sustancias como la vitronectina, el factor de von Willebrand y la fibronectina, pero el fibrinógeno es el principal ligando debido a que contiene una mayor concentración de esta secuencia de aminoácidos. El diseño de agentes capaces de inhibir los receptores de GPIIb/IIIa permite el bloqueo de la etapa final del proceso trombótico, cualquiera que sea el mecanismo o la sustancia que inicialmente lo activara. Esto convierte a estos receptores en la diana ideal para el tratamiento de los síndromes coronarios agudos37. Con este objetivo, se han diseñado miles de compuestos, la mayoría de los cuales no ha progresado a fase de ensayo clínico. Por su mecanismo de acción, se puede considerar dos tipos o familias de fármacos antagonistas de la GPIIb/IIIa plaquetaria: los que bloquean de manera permanente los receptores plaquetarios (abciximab) y los que los inhiben de manera competitiva y reversible, el lugar de unión para la secuencia RGD, cuyo efecto depende de la concentración plasmática (eptifibatida, tirofibán y lamifibán) (fig. 1)38.
Los cuatro fármacos se administran por vía endovenosa; se han demostrado eficaces en ensayos clínicos a gran escala y están en actual uso clínico39. Se han desarrollado otros fármacos endovenosos inhibidores de la GPIIb/IIIa, incluidos el YM337 y el fradafibán, pero nunca han sido aprobados para uso clínico. Igualmente, se han desarrollado varios fármacos inhibidores orales de la GPIIb/IIIa para el tratamiento a largo plazo de pacientes con enfermedad coronaria estable. En contraste con los beneficios asociados a la administración endovenosa de antagonistas del receptor IIb/IIIa, los resultados obtenidos tras la administración de antagonistas orales (p. ej., xemilofibán, sibrafibán, orbofibán) fueron desalentadores. De hecho, diversos ensayos clínicos a gran escala han demostrado un aumento de la mortalidad tras el uso de varios preparados orales que, paradójicamente, se asociaron a más trombosis.
Antagonistas del receptor de la trombinaLa trombina no es sólo un componente esencial en la cascada de coagulación, sino que además es primordial en la evolución del proceso aterotrombótico40. La trombina activa las plaquetas al interactuar con dos receptores acoplados a proteínas G (fig. 2) pertenecientes a la familia de los receptores activados por proteasas, el PAR1 y el PAR4, y en menor grado con la GPIbα42. PAR-1 presenta gran afinidad por la trombina y por ello es el efector principal de la cascada de señalización derivada de la interacción trombina-plaqueta. PAR-4 presenta una menor afinidad y junto a la GPIbα complementa a PAR-1 en las fases más tardías del proceso de activación. La activación del receptor se lleva a cabo mediante un proceso de proteolisis, a saber, la unión de la trombina con PAR produce una escisión del segmento aminoterminal del receptor y se generan nuevas cadenas aminoacídicas terminales (SFLLRN para PAR-1 y GYPGKF para PAR-4) que posteriormente autoestimulan otras regiones del receptor42. Hasta la fecha se han evaluado a nivel preclínico cuatro antagonistas distintos del PAR-1 (SCH530348, SCH205831, SCH602539 y E5555) aunque sólo de dos, el SCH530348 (vorapaxar) y el E5555 (atopaxar), se ha realizado una investigación más avanzada en un ensayo clínico en fase III (figs. 1 y2).
El vorapaxar y el atopaxar son moléculas no proteicas que actúan como potentes antagonistas competitivos del receptor PAR-1 de la trombina. Ambos se administran por vía oral. El vorapaxar se absorbe rápidamente y consigue una rápida acción antiplaquetaria a las 2h, mientras que el atopaxar es algo más tardío (3,5h). El vorapaxar presenta una larga semivida, estimada de hasta 311h, mientras que la del atopaxar es de 23h. Ambos presentan una metabolización hepática lenta por el citocromo P450 CYP3A4. Por lo tanto, la administración concomitante de fármacos que modifican la actividad metabólica de la enzima (p. ej., ketoconazol, rifampicina) puede modular la concentración final de vorapaxar. La eliminación de ambos fármacos es principalmente vía fecal (95%), aunque un 5% se excreta vía renal.
Hasta la fecha se han llevado a cabo ensayos clínicos en fase III43,44 en los que se ha visto que la adición de vorapaxar a AAS y clopidogrel no mejora los objetivos principal ni secundario e incrementa significativamente el sangrado mayor y la hemorragia cerebral.
Moduladores del ampc plaquetarioLa activación y la agregación plaquetarias también pueden detenerse por elevación de la concentración de AMPc intracitoplásmico, lo cual se acompaña de una disminución del calcio intracitoplásmico, por transporte al interior de los gránulos densos, lo que causa inhibición de la función plaquetaria (fig. 2). La concentración intraplaquetaria de AMPc se regula por el sistema de la adenilciclasa (enzimas de membrana que catalizan la conversión del ATP en AMPc) y por las fosfodiesterasas, que catalizan el catabolismo del AMPc a AMP. Tanto los fármacos que estimulan la acción de la adenilciclasa como los que inhiben la acción de la fosfodiesterasa pueden tener acción antiagregante plaquetaria. Entre los primeros se encuentra la prostaciclina y entre los segundos, el dipiridamol (fig. 1). El dipiridamol inhibe la captación de adenosina en los eritrocitos, las plaquetas y las células endoteliales in vitro e in vivo. Por consiguiente, se produce un aumento de la concentración de adenosina localmente que actúa sobre el receptor A2 plaquetario, estimula la adenilciclasa plaquetaria y, por lo tanto, aumenta la concentración de AMPc plaquetario (fig. 2). Por ello, se inhibe la agregación plaquetaria en respuesta a diversos estímulos tales como PAF, colágeno y ADP y presenta propiedades vasodilatadoras.
Futuras Aproximaciones AntiplaquetariasA pesar del gran avance en el arsenal antiplaquetario para el tratamiento y la prevención de la enfermedad isquémica coronaria45 y la enfermedad cerebrovascular46, estos fármacos presentan aún ciertas limitaciones que dejan sin proteger a ciertos grupos de pacientes. Por ello, los esfuerzos actuales se centran no sólo en perfeccionar los agentes ya en uso a fin de que estos aumenten su eficacia y su seguridad, sino en desarrollar nuevos agentes que, mediante el bloqueo de nuevas dianas, se presenten como atractivas alternativas terapéuticas o aditivos a los regímenes antiplaquetarios ya establecidos sin aumentar el riesgo hemorrágico. Es necesario seguir profundizando en un mejor conocimiento de la fisiopatología de la activación plaquetaria. El notorio progreso en genómica y proteómica está generando información relevante acerca de los procesos que integran y vinculan las interacciones plaqueta-agonista, con las consiguientes vías de señalización y posterior estabilización del trombo, con el fin de desarrollar nuevas dianas terapéuticas que inhiban selectivamente los procesos desencadenantes de la enfermedad aterotrombótica.
FinanciaciónEste artículo ha sido posible gracias a la financiación de PNSAF2010-16549, CIBERobn-Instituto de Salud Carlos III, TerCel RD06/0010/0017 y Fundación Jesús Serra. GV es RyC contratada (RyC-2009-5495) del MICINN.
Conflicto de interesesNinguno.