Se han producido avances importantes en el campo de la genética molecular que han ampliado nuestra capacidad de identificar sustratos genéticos subyacentes a la patogenia de diversos trastornos que siguen patrones de herencia mendeliana. Entre estos trastornos, se encuentran las canalopatías y miocardiopatías hereditarias y potencialmente mortales con base genética subyacente identificada y que ahora se conoce mejor. La heterogeneidad clínica y genética es una característica distintiva de estos trastornos, con miles de mutaciones génicas involucradas en estas enfermedades cardiovasculares divergentes. Las pruebas genéticas en varias de estas canalopatías y miocardiopatías hereditarias han alcanzado ya la fase de madurez, tras evolucionar desde su descubrimiento a las pruebas genéticas para investigación y las pruebas diagnósticas disponibles para uso clínico/comercial. El objetivo de esta revisión es proporcionar al lector un conocimiento básico de la genética médica humana y las pruebas genéticas existentes en el contexto de las enfermedades cardiovasculares que afectan al corazón. Revisamos el estado actual de las pruebas genéticas clínicas para las canalopatías y miocardiopatías más frecuentes, tratamos asuntos pertinentes que surgen de utilizar pruebas genéticas y sobre el futuro de la medicina personalizada en las enfermedades cardiovasculares.
Palabras clave
Los considerables avances que se han producido en el campo de la genética molecular han aportado instrumentos importantes para esclarecer los sustratos genéticos de muchos trastornos genéticos que siguen patrones de herencia mendelianos. Se ha identificado y actualmente se conoce mejor el fundamento genético de ciertas miocardiopatías y canalopatías cardiacas hereditarias y potencialmente mortales, como la miocardiopatía hipertrófica (MCH), la miocardiopatía dilatada (MCD), la displasia arritmogénica de ventrículo derecho, el síndrome de QT largo (LQTS), la taquicardia ventricular polimórfica catecolaminérgica (CPVT) y el síndrome de Brugada (SB).
La notable heterogeneidad clínica y genética es una característica clave de estos trastornos, con múltiples genes subyacentes en sus mecanismos fisiopatológicos. Las pruebas genéticas para varias de estas miocardiopatías y canalopatías hereditarias han completado ya el paso del descubrimiento en el ámbito de la investigación a una aplicación con utilidad clínica y están ahora disponibles en el mercado como pruebas genéticas clínicas.
En esta revisión, se evalúa la situación actual de las pruebas genéticas clínicas para las canalopatías/miocardiopatías más frecuentes, es decir: LQTS, CPVT, SB y MCH. A continuación se abordan la necesidad de una interpretación cuidadosa de los resultados de las pruebas genéticas, la importancia del consejo genético y el futuro de la medicina personalizada en la enfermedad cardiovascular.
PRUEBAS GENÉTICAS PARA MIOCARDIOPATÍAS Y CANALOPATÍAS CARDIACAS HEREDITARIASAunque las pruebas genéticas para la MCD y la displasia arritmogénica de ventrículo derecho están evolucionando rápidamente y se comercializan, la interpretación de los resultados de las pruebas resulta extremadamente difícil, dado que no está clara su repercusión clínica, excepto en las pruebas que determinan mutaciones para la confirmación en familiares de primer grado, así como por su bajo rendimiento y el «ruido de fondo» comparativamente elevado1,2. Esta revisión se centra en el LQTS, la CPVT, el SB y la MCH, en los que la utilidad clínica de las pruebas genéticas hoy se puede decir que es máxima. En los apartados que siguen, dedicados a cada una de estas enfermedades, se presenta una breve descripción clínica, la base genética del trastorno y las recomendaciones actuales del consenso de expertos de Heart Rhythm Society (HRS)/European Heart Rhythm Association (EHRA) para las pruebas genéticas3.
Síndrome de QT largoDescripción clínicaEl LQTS se caracteriza por un retraso en la repolarización del miocardio, prolongación del intervalo QT y anomalías de la onda T en el electrocardiograma (ECG) de superficie de 12 derivaciones obtenido en reposo, lo cual puede manifestarse en forma de síncope, crisis convulsivas y muerte súbita cardiaca en pacientes con corazón estructuralmente normal4. Aunque la prevalencia del LQTS se estima en hasta 1:2.500 personas, debido a la heterogeneidad de esta enfermedad, se puede no detectar a los sujetos con LQTS, que pueden no mostrar prolongación del intervalo QT en el ECG de 12 derivaciones en reposo5. Las manifestaciones típicas de síncope, crisis convulsivas y muerte súbita cardiaca suelen darse tras la estimulación adrenérgica (estímulos auditivos, ejercicio, emoción, etc.) o durante el puerperio6. La arritmia característica distintiva de torsade de pointes con frecuencia se revierte espontáneamente a ritmo sinusal normal y causa sólo un episodio de síncope; sin embargo, aproximadamente un 5% de las personas no tratadas y sin sospecha de LQTS fallecen por una arritmia mortal como primer episodio clínico. Muchos de estos sujetos pueden haber sufrido algún episodio cardiaco previo no reconocido compatible con el fenotipo de LQTS. Además, aproximadamente un 20% de las muertes súbitas con autopsia negativa no explicadas de sujetos jóvenes y un 10% de las muertes por síndrome de muerte súbita infantil pueden ser muertes desencadenadas por LQTS7,8.
Base genéticaEl LQTS, descrito anteriormente como síndrome de Romano-Ward, es un trastorno genéticamente heterogéneo que la mayor parte de las veces se hereda de manera autosómica dominante. La forma recesiva del LQTS, descrita anteriormente como síndrome de Jervell y de Lange-Nielson, se caracteriza por un fenotipo cardiaco grave y pérdida de audición neurosensitiva. Hoy están identificadas centenares de mutaciones en 10 genes de susceptibilidad al LQTS (tabla 1) que originan un fenotipo de LQTS «clásico» no sindrómico. Además, hay tres trastornos multisistémicos muy poco frecuentes (síndrome de anquirina B, síndrome de Anderson-Tawil y síndrome de Timothy), anteriormente designadas como LQT4, LQT7 y LQT8, respectivamente9–11. También se han descrito mutaciones muy poco frecuentes aparecidas de novo, en vez de las de línea germinal familiares, que explican aproximadamente un 5-10% de los casos de LQTS.
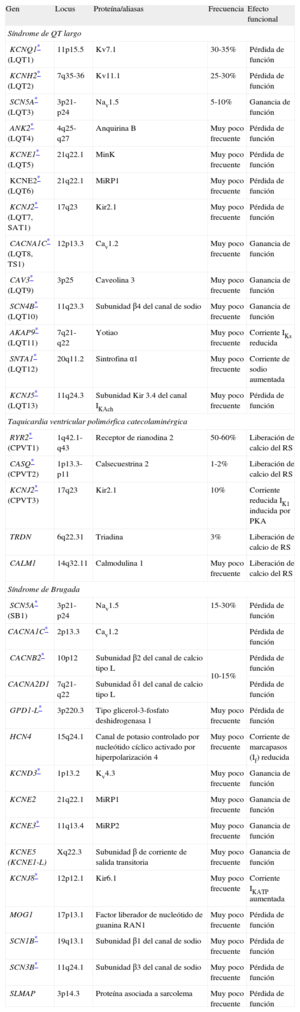
Resumen de los genes asociados a canalopatías cardiacas hereditarias
| Gen | Locus | Proteína/aliasas | Frecuencia | Efecto funcional |
| Síndrome de QT largo | ||||
| KCNQ1* (LQT1) | 11p15.5 | Kv7.1 | 30-35% | Pérdida de función |
| KCNH2* (LQT2) | 7q35-36 | Kv11.1 | 25-30% | Pérdida de función |
| SCN5A* (LQT3) | 3p21-p24 | Nav1.5 | 5-10% | Ganancia de función |
| ANK2* (LQT4) | 4q25-q27 | Anquirina B | Muy poco frecuente | Pérdida de función |
| KCNE1* (LQT5) | 21q22.1 | MinK | Muy poco frecuente | Pérdida de función |
| KCNE2* (LQT6) | 21q22.1 | MiRP1 | Muy poco frecuente | Pérdida de función |
| KCNJ2* (LQT7, SAT1) | 17q23 | Kir2.1 | Muy poco frecuente | Pérdida de función |
| CACNA1C* (LQT8, TS1) | 12p13.3 | Cav1.2 | Muy poco frecuente | Ganancia de función |
| CAV3* (LQT9) | 3p25 | Caveolina 3 | Muy poco frecuente | Ganancia de función |
| SCN4B* (LQT10) | 11q23.3 | Subunidad β4 del canal de sodio | Muy poco frecuente | Ganancia de función |
| AKAP9* (LQT11) | 7q21-q22 | Yotiao | Muy poco frecuente | Corriente IKs reducida |
| SNTA1* (LQT12) | 20q11.2 | Sintrofina α1 | Muy poco frecuente | Corriente de sodio aumentada |
| KCNJ5* (LQT13) | 11q24.3 | Subunidad Kir 3.4 del canal IKAch | Muy poco frecuente | Pérdida de función |
| Taquicardia ventricular polimórfica catecolaminérgica | ||||
| RYR2* (CPVT1) | 1q42.1-q43 | Receptor de rianodina 2 | 50-60% | Liberación de calcio del RS |
| CASQ* (CPVT2) | 1p13.3-p11 | Calsecuestrina 2 | 1-2% | Liberación de calcio del RS |
| KCNJ2* (CPVT3) | 17q23 | Kir2.1 | 10% | Corriente reducida IK1 inducida por PKA |
| TRDN | 6q22.31 | Triadina | 3% | Liberación de calcio de RS |
| CALM1 | 14q32.11 | Calmodulina 1 | Muy poco frecuente | Liberación de calcio del RS |
| Síndrome de Brugada | ||||
| SCN5A* (SB1) | 3p21-p24 | Nav1.5 | 15-30% | Pérdida de función |
| CACNA1C* | 2p13.3 | Cav1.2 | Pérdida de función | |
| CACNB2* | 10p12 | Subunidad β2 del canal de calcio tipo L | 10-15% | Pérdida de función |
| CACNA2D1 | 7q21-q22 | Subunidad δ1 del canal de calcio tipo L | Pérdida de función | |
| GPD1-L* | 3p220.3 | Tipo glicerol-3-fosfato deshidrogenasa 1 | Muy poco frecuente | Pérdida de función |
| HCN4 | 15q24.1 | Canal de potasio controlado por nucleótido cíclico activado por hiperpolarización 4 | Muy poco frecuente | Corriente de marcapasos (If) reducida |
| KCND3* | 1p13.2 | Kv4.3 | Muy poco frecuente | Ganancia de función |
| KCNE2 | 21q22.1 | MiRP1 | Muy poco frecuente | Ganancia de función |
| KCNE3* | 11q13.4 | MiRP2 | Muy poco frecuente | Ganancia de función |
| KCNE5 (KCNE1-L) | Xq22.3 | Subunidad β de corriente de salida transitoria | Muy poco frecuente | Ganancia de función |
| KCNJ8* | 12p12.1 | Kir6.1 | Muy poco frecuente | Corriente IKATP aumentada |
| MOG1 | 17p13.1 | Factor liberador de nucleótido de guanina RAN1 | Muy poco frecuente | Pérdida de función |
| SCN1B* | 19q13.1 | Subunidad β1 del canal de sodio | Muy poco frecuente | Pérdida de función |
| SCN3B* | 11q24.1 | Subunidad β3 del canal de sodio | Muy poco frecuente | Pérdida de función |
| SLMAP | 3p14.3 | Proteína asociada a sarcolema | Muy poco frecuente | Pérdida de función |
CPVT: taquicardia ventricular polimórfica catecolaminérgica; LQT: QT largo; RS: retículo sarcoplásmico; SAT: síndrome de Anderson-Tawil; SB: síndrome de Brugada; TS: síndrome de Timothy.
Genes con pruebas genéticas disponibles en el mercado. Muy poco frecuente se define por el hecho de explicar menos del 1% de la enfermedad. Obsérvese que los genotipos que tienen designación numérica se indican en orden ascendente. Los genes de susceptibilidad a la enfermedad, sin designación numérica, se indican en orden alfabético.
Aproximadamente un 75% de los LQTS se deben a mutaciones de tres genes —KCNQ1 (LQT1), KCNH2 (LQT2) y SCN5A (LQT3)— que contienen el código de las subunidades α formadoras de poros del canal iónico de potasio (Kv7.1 y Kv11.1) o de sodio (Nav1.5) y son cruciales para modular el potencial de acción cardiaco de los miocitos ventriculares12,13. El resto de los casos de LQTS con genotipo positivo se atribuyen a mutaciones de genes que contienen el código de otros canales iónicos cardiacos o de proteínas que interaccionan con los canales iónicos y regulan la función de estos. Los siete genes menores de susceptibilidad al LQTS explican menos del 5% de los casos de LQTS.
Hasta la fecha, la mayoría de las mutaciones descritas en el LQTS corresponden a sustituciones de un solo nucleótido e inserciones/deleciones pequeñas situadas dentro de regiones de codificación de los genes de susceptibilidad al LQTS. Sin embargo, en el LQTS se han descrito también unos pocos ejemplos de reordenaciones génicas amplias que causan grandes deleciones/duplicaciones génicas14. Aunque se han realizado grandes avances en el conocimiento de la etiología subyacente al LQTS y se han descrito ya 10 genes, es importante comprender que en aproximadamente un 20-25% de los LQTS se continúa sin esclarecer un origen genético. Con los recientes avances de los métodos moleculares, el estudio continuo de pacientes con un fenotipo claro y un genotipo negativo es lo que brinda la mayor probabilidad de identificar el fundamento genético del 20-25% restante de LQTS.
Se han descrito unas pocas correlaciones de genotipo-fenotipo en pacientes con LQT1, LQT2 y LQT3 que pueden ser útiles para orientar las pruebas genéticas del LQTS. En el LQT1, los episodios cardiacos más frecuentes se observan a menudo durante el ejercicio y nadando; en cambio, en el LQT2 y el LQT3, los episodios cardiacos se dan con mayor frecuencia durante periodos de reposo/sueño. Los episodios debidos a un estímulo auditivo y la susceptibilidad de las mujeres durante el puerperio son característicos del LQT26,15. Sin embargo, cuando se produce un síncope inducido por el ejercicio en un paciente con un QTc normal (< 440 ms los varones y < 460 ms las mujeres), este fenotipo puede indicar una CPVT16.
Se han descrito también patrones electrocardiográficos que apuntan a un gen alterado6,15 y pueden ser útiles para orientar las pruebas genéticas. Por ejemplo, las ondas T de base ancha se asocian al LQT1, las ondas T de baja amplitud con una escotadura o bifásicas se asocian al LQT2 y un segmento isoeléctrico largo seguido de una onda T de base estrecha se asocia al LQT3. Sin embargo, estos patrones de onda T que apuntan a un gen solamente indican el subtipo de LQTS y no se debe usarlos para una predicción previa a la prueba genética. Este abordaje cauteloso es de capital importancia, ya que la base genética subyacente no sólo tiene consecuencias diagnósticas, sino que puede tener gran influencia en las recomendaciones terapéuticas en el LQTS. Un ejemplo de ello se puede observar en la respuesta de los pacientes con LQTS a los bloqueadores beta, el tratamiento farmacológico estándar del LQTS. Hemos observado que los bloqueadores beta aportan la máxima protección a los pacientes con LQT1, una protección moderada a los pacientes con LQT2 y posiblemente no aportan protección suficiente a los pacientes con LQT317,18. Sin embargo, el tratamiento dirigido a la corriente de sodio tardía, que es característica de las mutaciones de LQT3, con fármacos como mexiletina, flecainida, ranolazina o propranolol puede ser una terapia génica más específica para los pacientes con LQT36,15,19,20.
Se ha realizado una estratificación intragenotípica para el LQT1 y el LQT2, basada en el tipo de mutación, la localización de esta y la función celular21,22. Los pacientes con mutaciones de cambio de sentido de LQT1 que se localizan en regiones transmembranarias del canal de potasio Kv7.1 tienen el doble de riesgo de un episodio cardiaco desencadenado por LQT1 que los pacientes con LQT1 con mutaciones localizadas en el extremo carboxílico. Además, los estudios celulares in vitro de las mutaciones de pacientes con LQT1 que produjeron un fenotipo con mayor pérdida de función (es decir, dominante negativo) muestran el doble de riesgo clínico que las mutaciones con menos impacto en la biología del canal Kv7.1 (es decir, haploinsuficiencia). Al igual que en la estratificación molecular del riesgo observada en los pacientes con LQT1, los pacientes con LQT2 tienen también factores de riesgo independientes relacionados con la localización de la mutación y la función celular. Los pacientes con LQT2 con mutaciones de la región del poro de Kv11.1 tienden a presentar un QTc más largo, sufren mayor número de eventos cardiacos por arritmias a edad más temprana y tienen una manifestación clínica del trastorno más grave que la de los pacientes con LQT2 y mutaciones no localizadas en la región del poro del canal Kv11.1. Además, estudios recientes han demostrado que las mutaciones de Kv11.1 de LQT2 que afectan a la región del poro transmembranario son las que tienen mayor riesgo de eventos cardiacos, las que corresponden a mutaciones de desplazamiento de marco de lectura/mutaciones sin sentido tienen un riesgo intermedio, y las que consisten en mutaciones de cambio de sentido en la región carboxiterminal son las que tienen el riesgo más bajo de eventos cardiacos22. En 2012, la determinación del genotipo del LQTS cumple plenamente la tríada por lo que respecta al impacto diagnóstico (aproximación/alejamiento del diagnóstico en el caso índice, verificación/exclusión del patrón de referencia en familiares), pronóstico (tanto intergenotípico como intragenotípico) y terapéutico (p. ej., se aborda a los pacientes con LQT1 con un plan de tratamiento fundamentalmente distinto del de los pacientes con LQT3).
Recomendaciones de pruebas genéticas para el síndrome de QT largoSe debe ofrecer pruebas genéticas clínicas a todo paciente con diagnóstico de sospecha clínica de LQTS. Como se ha descrito antes, la determinación del genotipo proporciona una estratificación adicional del riesgo que puede orientar la elección de las opciones terapéuticas de las que dispone el clínico. Las pruebas genéticas en el caso índice tienen actualmente una probabilidad del 75% de identificar una mutación asociada al LQTS posible/probable. Si el caso índice presenta un resultado positivo de las pruebas genéticas, la determinación del genotipo de los familiares inmediatos permite perfeccionar el diagnóstico genético, con independencia de los síntomas o el intervalo QT corregido. Además, en los pacientes con síncope de esfuerzo no explicado o una prolongación del intervalo QT/torsade de pointes inducida por fármacos, pero que no cumplen por completo los criterios diagnósticos del LQTS y se les asigna un diagnóstico de LQTS «posible», puede ser útil también la realización de pruebas genéticas del LQTS. En esos casos, un resultado positivo de las pruebas genéticas puede ser útil para pasar del diagnóstico clínicamente «posible» al de genéticamente probable. En cambio, un resultado negativo de las pruebas genéticas permite descartar el 75% de los LQTS congénitos y aporta una medida independiente que ayuda al clínico a apartarse de este diagnóstico, sobre todo si el fenotipo clínico es débil.
En consecuencia, las recomendaciones del consenso de expertos de HRS/EHRA3 para la realización de pruebas genéticas del LQTS son: a) las pruebas genéticas del LQTS completas o dirigidas a LQT1-3 (KCNQ1, KCNH2 y SCN5A) se recomiendan para cualquier paciente en el que un cardiólogo haya establecido un alto grado de sospecha clínica de LQTS en función del examen de los antecedentes clínicos del paciente, los antecedentes familiares y el fenotipo expresado en el ECG (ECG de 12 derivaciones en reposo o pruebas de estrés de provocación con ejercicio o con infusión de catecolaminas); b) las pruebas genéticas del LQTS completas o dirigidas a LQT1-3 (KCNQ1, KCNH2 y SCN5A) se recomiendan para cualquier paciente asintomático con prolongación del intervalo QT y sin otros trastornos clínicos que pudieran prolongarlo (como anomalías electrolíticas, hipertrofia, bloqueo de rama del haz, etc.; es decir, idiopática) en los ECG de 12 derivaciones secuenciales, y definida como un QTc > 480 ms (antes de la pubertad) o > 500 ms (en el adulto); c) se puede considerar las pruebas genéticas del LQTS completas o dirigidas a LQT1-3 (KCNQ1, KCNH2 y SCN5A) para cualquier paciente asintomático con unos valores de QTc > 460 ms (antes de la pubertad) o > 480 ms (en el adulto) en los ECG de 12 derivaciones seriados, por lo demás idiopáticos, y d) las pruebas genéticas específicas para determinada mutación se recomiendan para los familiares y otros parientes apropiados tras identificar la mutación causante del LQTS en un caso índice.
Taquicardia ventricular polimórfica catecolaminérgicaDescripción clínicaLa CPVT es un síndrome de arritmia hereditaria potencialmente mortal, que se expresa principalmente en sujetos jóvenes y suele manifestarse por síncope, crisis convulsivas o muerte súbita asociadas al ejercicio. Anteriormente se pensaba que se manifestaba únicamente durante la infancia, pero estudios recientes han señalado que este síndrome puede presentarse por primera vez a cualquier edad desde el lactante hasta los 40 años. Al igual que ocurre con el LQT1, nadar es un desencadenante típico de eventos arritmogénicos en la CPVT. De hecho, tanto la CPVT como el LQT1 son causa de muchos casos de casi ahogamiento o ahogamiento inexplicado de nadadores por lo demás sanos23. Sin embargo, contrariamente a lo que ocurre en el LQTS, la CPVT se asocia a un ECG de 12 derivaciones en reposo completamente normal (aunque a veces con bradicardia y ondas U), pero puede producir una extrasistolia ventricular importante, con TV bidireccional después de una prueba de estrés en ejercicio o con catecolaminas24. La letalidad de la CPVT se pone de manifiesto en unas tasas de mortalidad del 30 al 50% al llegar a los 35 años y por la presencia de unos antecedentes familiares positivos de muerte súbita cardiaca de individuos jóvenes (< 40 años) en más de una tercera parte de los pacientes con CPVT y hasta un 60% de las familias en que hay mutaciones de RyR225. Además, la CPVT es la base patogénica que subyace a aproximadamente un 15% de las muertes súbitas inexplicadas de sujetos jóvenes con autopsia negativa8.
Base genéticaLa CPVT suele heredarse en forma de un trastorno autosómico dominante. En la patogenia de la CPVT subyacen alteraciones de la liberación de calcio intracelular a partir del retículo sarcoplásmico. Las mutaciones de ganancia de función del receptor de rianodina cardiaca codificada en el RYR2 explican la mayoría de los casos (∼60%) de CPVT. Estas mutaciones de RyR2 causan una liberación diastólica excesiva de calcio del retículo sarcoplásmico, lo cual puede conducir a un retraso de las posdespolarizaciones y arritmias ventriculares25. La inmensa mayoría (90%) de las mutaciones de RYR2 son de cambio de sentido; sin embargo, hasta un 5% de los pacientes con CPVT no relacionados son portadores de amplias reordenaciones de RYR2 que causan deleciones grandes del exón. Se han identificado dos formas recesivas de CPVT que se deben a mutaciones en la proteína calsecuestrina 2 codificada en CASQ2 y de la triadina codificada en TRDN, dos proteínas con interacción con RYR226,27. Muy recientemente se han identificado mutaciones en la calmodulina 1 (CALM1) como causa de una CPVT autosómica dominante, y se ha identificado una única mutación de cambio de sentido segregada con un fenotipo de CPVT en la genealogía de una familia amplia de Suecia (tabla 1)28.
Es importante señalar que casi una tercera parte de los casos de LQTS «posible/atípico» (QTc < 480 ms) con síncope o parada cardiaca relacionados con ejercicio se han identificado como casos positivos de mutación de RYR2. Así pues, la manifestación clínica de eventos cardiacos (es decir, síncope, crisis convulsivas o parada cardiaca) inducidos por el ejercicio y QTc < 460 ms debe hacer pensar siempre en una CPVT como primera consideración clínica, más que en un «LQT1 oculto». De hecho, hasta un 30% de los pacientes con CPVT pueden haber sido diagnosticados erróneamente de «LQTS con intervalos QT normales» o «LQTS oculto», lo cual ilustra la enorme trascendencia clínica que tiene la distinción adecuada entre CPVT y LQTS, puesto que las evaluaciones del riesgo y las estrategias de tratamiento de estos diferentes trastornos pueden ser distintas. De igual modo, se sabe que algunos pacientes a los que se ha diagnosticado una CPVT con presencia de una TV bidireccional con el ejercicio albergan mutaciones de KCNJ2 relacionadas con el síndrome de Andersen-Tawil tipo 1, que rara vez es mortal29. Un síndrome de Anderson-Tawil mal diagnosticado como CPVT, un trastorno de alta letalidad, podría conducir a un tratamiento profiláctico más agresivo de lo necesario (es decir, implante de desfibrilador automático implantable). Las pruebas genéticas pueden aportar una diferenciación diagnóstica clara entre CPVT y LQT1 oculto y entre CPVT y síndrome de Anderson-Tawil tipo 1.
Recomendaciones para las pruebas genéticas en la taquicardia ventricular polimórfica catecolaminérgicaLas recomendaciones del consenso de expertos de HRS/EHRA3 para las pruebas genéticas en la CPVT establecen: a) las pruebas genéticas de la CPVT completas o dirigidas a CPVT1 y CVPT2 (RYR2 y CASQ2) se recomiendan para cualquier paciente en el que un cardiólogo haya establecido cierto grado de sospecha clínica de una CPVT en función del examen de los antecedentes clínicos del paciente, los antecedentes familiares y el fenotipo electrocardiográfico expresado durante las pruebas de provocación de estrés en bicicleta, cinta sin fin o infusión de catecolaminas, y b) las pruebas genéticas específicas para determinada mutación se recomiendan para los familiares y parientes apropiados tras identificar la mutación causante de la CPVT en un caso índice. Además, para los pacientes con síncope/parada cardiaca inducidos por el ejercicio o casi ahogamiento en el contexto de un QTc < 460 ms, se debe considerar la realización de pruebas genéticas para la CPVT.
Síndrome de BrugadaDescripción clínicaEl SB es un síndrome hereditario potencialmente mortal que se caracteriza por una elevación del segmento ST con morfología descendente (≥ 2 mm) seguida de una onda T negativa registrada en las derivaciones precordiales derechas V1-V3 (patrón electrocardiográfico de SB «tipo 1»), y un aumento de la predisposición a la muerte súbita como consecuencia de episodios de TV polimórfica. Se ha observado también un patrón electrocardiográfico «tipo 2», consistente en una morfología del segmento ST en silla de montar ≥ 2 mm seguido de una onda T positiva, y un patrón electrocardiográfico «tipo 3», consistente en una elevación del segmento ST con morfología descendente o en silla de montar ≤ 1 mm, que no se considera diagnóstico de SB. Aunque el SB puede manifestarse a cualquier edad, afecta predominantemente a varones adultos jóvenes (30-40 años). Los síntomas, incluida la muerte súbita cardiaca, suelen producirse durante el sueño30.
Base genéticaEl SB se hereda habitualmente con un patrón autosómico dominante, aunque hasta la mitad de los casos pueden ser de carácter esporádico. Hasta la fecha, se han identificado 15 genes de susceptibilidad al SB (tabla 1). Las mutaciones de pérdida de función en la subunidad α del canal de sodio cardiaco codificado en SCN5A constituyen el sustrato genético más frecuente del SB y explican un 15-30% de los casos del trastorno31. Aparte de la disfunción del canal de sodio, las mutaciones que afectan al canal de calcio de tipo L en las subunidades α1(CACNA1C)32, β2(CACNB2B)32 y α2δ (CACNA2D1)33 pueden causar un 10-15% de los casos de SB, especialmente cuando se acompañan de intervalos QT breves33. Sin embargo, en 2012 Crotti et al. llevaron a cabo el primer análisis mutacional amplio en una cohorte grande de pacientes con SB no emparentados. Aunque identificaron mutaciones de SCN5A en un 16% de los pacientes de su cohorte, sólo un 1,5% de sus casos de SB tenían una mutación en uno de los tres genes de las subunidades del canal de calcio tipo L en ausencia de intervalo QT corto. De hecho, todos los genes menores del SB suponen en conjunto menos del 5% de los casos del trastorno31. Así pues, es importante tener en cuenta que, en 2012, aproximadamente un 70-80% de los casos de SB continúan sin esclarecerse genéticamente.
Aunque son algo limitadas, hay algunas correlaciones de genotipo/fenotipo clave derivadas de las pruebas genéticas realizadas en el estudio de pacientes con SB que pueden ser útiles para orientar los estudios futuros de pruebas genéticas y aportar cierta utilidad pronóstica a las pruebas genéticas del SB. Por ejemplo, la presencia de un intervalo PQ largo (≥ 200 ms) en el ECG indica en mayor medida un SB producido a través de SCN5A34. De hecho, Crotti et al. han descrito que, en comparación con un rendimiento < 10% de un resultado positivo en la prueba genética de SCN5A para los pacientes con un PQ < 200 ms, el rendimiento fue de casi un 40% en los pacientes con intervalo PQ ≥ 200 ms. Además, la presencia de de intervalo QT corto (QTc 350 ms) apunta a un sustrato del SB en el canal de calcio de tipo L31.
En 2009, Meregalli et al. investigaron si el tipo de la mutación de SCN5A estaba correlacionado con la gravedad del retardo en la conducción presente en los fenotipos de ritmo aberrante de la enfermedad progresiva de la conducción cardiaca y del SB superpuestos35. Sus resultados pusieron de manifiesto que los pacientes con mutaciones que causaban mayor reducción del máximo de corriente de sodio tenían un fenotipo más grave del síndrome y defectos de la conducción, lo que constituye la primera estratificación del riesgo intragenotípica asociada a canalopatías del sodio con pérdida de función.
Recomendaciones de pruebas genéticas para el síndrome de BrugadaSe podría realizar pruebas genéticas clínicas a los pacientes con sospecha de SB siempre que se tenga presente que el rendimiento de las pruebas genéticas actualmente disponibles para el SB es de aproximadamente un 20-30%. Además, excepto para la reciente asociación entre el tipo de mutación de SCN5A y la gravedad clínica, el principal papel de las pruebas genéticas del SB es la confirmación diagnóstica de la causa básica de un caso índice. Ello se sigue de pruebas genéticas de confirmación a los familiares inmediatos, para diferenciar a los que requieren vigilancia clínica continua y medidas preventivas, como la evitación de ciertos fármacos (www.brugadadrugs.org)36, de aquellos que se puede considerar sin afección clínica, electrocardiográfica y genética.
Las recomendaciones del consenso de expertos de HRS/EHRA3 para las pruebas genéticas del SB son: a) las pruebas genéticas del SB completas o dirigidas al SB1 (SCN5A) pueden ser útiles para cualquier paciente en el que un cardiólogo haya establecido sospecha clínica de SB en función del examen de los antecedentes clínicos del paciente, los antecedentes familiares y el fenotipo expresado electrocardiográficamente (ECG de 12 derivaciones en reposo o pruebas de provocación con exposición a fármacos); b) las pruebas genéticas no están indicadas en el contexto de un patrón electrocardiográfico SB tipo 2 o 3 aislado, y c) se recomiendan pruebas genéticas específicas para determinada mutación para los familiares y parientes apropiados tras identificar la mutación causante del SB en un caso índice.
Miocardiopatía hipertróficaDescripción clínicaSe considera que la MCH es una hipertrofia ventricular izquierda asimétrica sin causa identificable clínicamente y con una prevalencia de 1:500 personas; es una causa frecuente de muerte súbita cardiaca entre los jóvenes, sobre todo deportistas. La expresividad de la MCH es muy variable y oscila entre un curso clínico asintomático durante toda la vida y dolor torácico, disnea de esfuerzo, síncope, intolerancia progresiva al ejercicio o muerte súbita en el primer episodio, que pueden darse a cualquier edad37,38. El fenotipo morfológico de la MCH va de la hipertrofia minúscula a la extensa, de muy poca fibrosis a una fibrosis extrema y desorden miocitario, y de ausencia de obstrucción infundibular del ventrículo izquierdo a obstrucción grave, con subtipos anatómicos específicos que incluyen la MCH de curva inversa, la MCH sigmoidea y la variante apical39.
Base genéticaLa MCH se hereda habitualmente en forma de trastorno autosómico dominante. Hasta la fecha se ha involucrado en el trastorno a por lo menos 27 presuntos genes de susceptibilidad a la MCH, con centenares de mutaciones identificadas (tabla 2). La MCH sarcomérica o miofilamentosa es el subtipo genético más frecuente de MCH, y se debe a mutaciones en genes que codifican proteínas de los miofilamentos gruesos y finos de la sarcómera cardiaca (tabla 2). Los dos genes más frecuentes asociados a la MCH, MYBPC3 y MYH7, tienen una prevalencia estimada de un 25-35% para cada gen y explican la mayor parte de los resultados positivos de las pruebas genéticas de investigación. Además, se ha relacionado con la patogenia de la MCH a los genes que codifican proteínas del disco Z cardiaco, de modulación del Ca2+ y reguladoras. Las mutaciones de varios genes que codifican proteínas relacionadas con el metabolismo celular dan lugar a trastornos específicos asociados a la hipertrofia ventricular izquierda que pueden semejarse al fenotipo de la MCH.
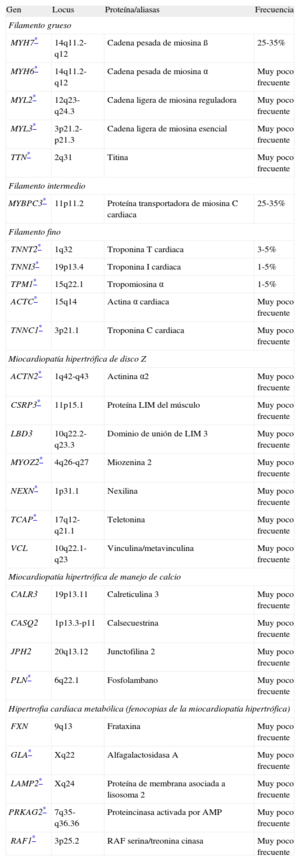
Resumen de los genes asociados a miocardiopatía hipertrófica
| Gen | Locus | Proteína/aliasas | Frecuencia |
| Filamento grueso | |||
| MYH7* | 14q11.2-q12 | Cadena pesada de miosina ß | 25-35% |
| MYH6* | 14q11.2-q12 | Cadena pesada de miosina α | Muy poco frecuente |
| MYL2* | 12q23-q24.3 | Cadena ligera de miosina reguladora | Muy poco frecuente |
| MYL3* | 3p21.2-p21.3 | Cadena ligera de miosina esencial | Muy poco frecuente |
| TTN* | 2q31 | Titina | Muy poco frecuente |
| Filamento intermedio | |||
| MYBPC3* | 11p11.2 | Proteína transportadora de miosina C cardiaca | 25-35% |
| Filamento fino | |||
| TNNT2* | 1q32 | Troponina T cardiaca | 3-5% |
| TNNI3* | 19p13.4 | Troponina I cardiaca | 1-5% |
| TPM1* | 15q22.1 | Tropomiosina α | 1-5% |
| ACTC* | 15q14 | Actina α cardiaca | Muy poco frecuente |
| TNNC1* | 3p21.1 | Troponina C cardiaca | Muy poco frecuente |
| Miocardiopatía hipertrófica de disco Z | |||
| ACTN2* | 1q42-q43 | Actinina α2 | Muy poco frecuente |
| CSRP3* | 11p15.1 | Proteína LIM del músculo | Muy poco frecuente |
| LBD3 | 10q22.2-q23.3 | Dominio de unión de LIM 3 | Muy poco frecuente |
| MYOZ2* | 4q26-q27 | Miozenina 2 | Muy poco frecuente |
| NEXN* | 1p31.1 | Nexilina | Muy poco frecuente |
| TCAP* | 17q12-q21.1 | Teletonina | Muy poco frecuente |
| VCL | 10q22.1-q23 | Vinculina/metavinculina | Muy poco frecuente |
| Miocardiopatía hipertrófica de manejo de calcio | |||
| CALR3 | 19p13.11 | Calreticulina 3 | Muy poco frecuente |
| CASQ2 | 1p13.3-p11 | Calsecuestrina | Muy poco frecuente |
| JPH2 | 20q13.12 | Junctofilina 2 | Muy poco frecuente |
| PLN* | 6q22.1 | Fosfolambano | Muy poco frecuente |
| Hipertrofia cardiaca metabólica (fenocopias de la miocardiopatía hipertrófica) | |||
| FXN | 9q13 | Frataxina | Muy poco frecuente |
| GLA* | Xq22 | Alfagalactosidasa A | Muy poco frecuente |
| LAMP2* | Xq24 | Proteína de membrana asociada a lisosoma 2 | Muy poco frecuente |
| PRKAG2* | 7q35-q36.36 | Proteincinasa activada por AMP | Muy poco frecuente |
| RAF1* | 3p25.2 | RAF serina/treonina cinasa | Muy poco frecuente |
Las pruebas genéticas de los ocho o nueve genes de susceptibilidad a la MCH establecidos para la MCH sarcomérica proporcionan un rendimiento de detección de mutaciones que oscila entre el 35 y el 65% en diversas cohortes de estudio internacionales formadas por pacientes no relacionados que cumplían el criterio clínico de MCH37,40. Además, las pruebas genéticas con guía ecográfica pueden ser útiles al clínico, pues proporcionan una estimación de la probabilidad a priori de una prueba genética positiva en función del subtipo morfológico establecido tras la ecocardiografía41.
Las pruebas genéticas de la MCH contribuyen principalmente a proporcionar la confirmación diagnóstica, y sólo tienen un papel menor en cuanto al pronóstico y las repercusiones terapéuticas. Las pruebas genéticas para la MCH pueden ser esenciales en la diferenciación entre una MCH verdadera y una hipertrofia por adaptación al ejercicio («corazón de atleta») o los trastornos que remedan el fenotipo de la MCH, como el síndrome de Anderson-Fabry, el almacenamiento de glucógeno (PRKAG2) y lisosoma (LAMP2), los síndromes mitocondriales y los síndromes de Noonan y LEOPARD, para los que existen tratamientos definitivos y alternativos que difieren de los de la MCH. Sin embargo, una prueba genética negativa no «descarta» la MCH. Además, los tratamientos profilácticos aportan la posibilidad de prevenir o retardar el inicio de la hipertrofia en los pacientes con genotipo positivo pero con resultado aún negativo en cuanto a hipertrofia.
Es preciso ser muy restrictivo al considerar la decisión de implantar un desfibrilador automático implantable basándose únicamente en el resultado de las pruebas genéticas. Actualmente, los datos clínicos existentes respaldan un indicador pronóstico de «efecto de clase de mutación», según el cual, en comparación con los pacientes con una MCH diagnosticada clínicamente y con una prueba genética negativa, los pacientes no relacionados con una prueba genética de MCH sarcomérica positiva expresan una hipertrofia más grave, una edad al diagnóstico más temprana y mayor probabilidad de progresión a enfermedad terminal42. Sin embargo, no está claro de qué forma puede trasladarse esta observación a un abordaje terapéutico del paciente con MCH sarcomérica/miofilamentosa (es decir, con una prueba genética positiva) en comparación con el paciente con MCH y prueba genética negativa.
Recomendaciones de pruebas genéticas para la miocardiopatía hipertróficaLas pruebas genéticas del paciente con MCH pueden aportar una referencia para el diagnóstico de sus familiares. El resultado positivo de una prueba genética podría permitir el análisis sistemático de los familiares de un caso inicial con MCH, con objeto de identificar a los familiares positivos para la mutación, con independencia de su manifestación fenotípica clínica en curso, lo que facilitaría la detección precoz y la selección adecuada de los familiares para la aplicación de vigilancia clínica continua, al tiempo que se descarta la necesidad de seguimiento con evaluaciones cardiacas y ecocardiografías regulares de los familiares con mutación negativa/fenotipo negativo.
En consecuencia, las recomendaciones del consenso de expertos de HRS/EHRA3 para las pruebas genéticas en la MCH establecen: a) las pruebas genéticas de la MCH completas o dirigidas (MYBPC3, MYH7, TNNI3, TNNT2, TPM1) se recomiendan para cualquier paciente en el que un cardiólogo haya establecido un diagnóstico clínico de MCH en función del examen de los antecedentes clínicos del paciente, los antecedentes familiares y el fenotipo electrocardiográfico/ecocardiográfico, y b) las pruebas genéticas específicas para determinada mutación se recomiendan para los familiares y parientes apropiados tras identificar la mutación causante de la MCH en un caso índice.
INTERPRETACIÓN DE LOS RESULTADOS DE LAS PRUEBAS GENÉTICASEl tratamiento clínico y la evaluación de los pacientes y las familias en las que se sospecha un trastorno cardiaco potencialmente mortal deben llevarse a cabo bajo la supervisión de un cardiólogo con conocimiento experto específico de las canalopatías y miocardiopatías hereditarias43. Dada la penetrancia incompleta y la expresividad variable asociadas a muchos de esos trastornos, la interpretación de los resultados de las pruebas genéticas debe realizarse con precaución, valorando cuidadosamente la asignación de patogenicidad a la variante genética identificada. Para ilustrar este punto cabe mencionar que, a diferencia de las mutaciones asociadas al LQTS que rara vez tienen efectos patogénicos y se estima que se dan en 1:2.500 personas (0,04%) y el 75% de los pacientes con LQTS con fenotipo clínico firme, un análisis mutacional completo de los tres genes principales del LQTS —KCNQ1 (LQT1), KCNH2 (LQT2) y SCN5A (LQT3)— en más de 1.300 voluntarios, por lo demás sanos, puso de relieve que aproximadamente un 4% de los caucásicos y hasta un 8% de los no caucásicos eran portadores de variantes genéticas infrecuentes (frecuencia alélica < 0,5%) con alteración de aminoácidos (no sinónimas) en estos genes de los canales cardiacos44. Aunque algunas de las variantes identificadas en esta población manifiestamente sana pueden corresponder a modificadores de enfermedad subclínica, la mayoría debe corresponder a un «ruido genético de fondo» benigno. Así pues, cuando se realizan a caucásicos con fenotipo firme, en comparación con los controles, las pruebas genéticas del LQTS tienen un cociente «señal/ruido» de aproximadamente 19:1. Esta observación de variantes de cambio de sentido no patogénicas de fondo no se limita al LQTS, sino que probablemente se extienda a todas las miocardiopatías y canalopatías cardiacas hereditarias que se detallan en esta revisión en diversos grados. Así pues, es de extraordinaria importancia reconocer que las pruebas genéticas de miocardiopatía/canalopatía son pruebas probabilísticas, más que binarias (sí o no). Para diferenciar las mutaciones causantes de enfermedad con efectos patogénicos de las variantes poco frecuentes de trascendencia incierta, están empezando a aparecer algoritmos de interpretación para cada uno de estos trastornos2,44.
CONSEJO GENÉTICOEl consejo genético es un componente esencial del proceso de realización de pruebas genéticas. Dada la complejidad de la información genética médica, resulta útil que el médico que las solicita colabore estrechamente con un asesor genético titulado y con formación de posgrado, que preferiblemente debería tener capacitación especializada en genética cardiovascular, formando un equipo que se comunique con el paciente en lo relativo a las implicaciones de las pruebas genéticas y sus beneficios y consecuencias. El consejo debe continuar durante todas las fases de aplicación de las pruebas, con objeto de proporcionar al paciente la información y los recursos necesarios para comprender la enfermedad genética, afrontarla y tomar decisiones al respecto45. Un asesor genético puede ser útil en muchos aspectos y puede facilitar la obtención de antecedentes familiares multigeneracionales completos y el aporte de información sobre el trastorno, su modo de transmisión hereditaria y sus implicaciones para la futura planificación familiar. Además, es importante que el asesor genético explique la prueba genética en sí, sus posibles limitaciones, sus costes y la posibilidad de resultados imprevistos, así como las posibles repercusiones de los resultados de las pruebas genéticas para el paciente y los familiares37. La National Society of Genetic Counselors (disponible en: www.nsgc.org)46 es un recurso útil para los clínicos que buscan a un asesor genético con experiencia en una enfermedad concreta.
PERSPECTIVAS FUTURAS EN EL DIAGNÓSTICO GENÉTICO CARDIOVASCULARSecuenciación completa de exoma/genoma de nueva generación para el diagnóstico genético sistemático y el descubrimiento de nuevos genes de enfermedadLa aparición de las plataformas de secuenciación de nueva generación que han reducido sustancialmente el coste de determinar el genotipo ha aportado nuevos instrumentos para investigar de manera eficiente el genoma o el exoma (región completa del genoma para codificación de aminoácidos) completo de un individuo con una sola reacción. En vez de abordar las pruebas genéticas con «un gen cada vez», los laboratorios especializados de genómica de todo el mundo están empezando a ofrecer la secuenciación de todo el genoma o de todo el exoma a partir de una muestra de ADN del paciente. Esta tecnología aporta de manera efectiva una lista de todas las sustituciones de nucleótido único y las inserciones/deleciones pequeñas (frecuentes o infrecuentes, benignas o patogénicas) de cada gen del genoma de un paciente.
La secuenciación de genoma completo o de exoma completo empleando la secuenciación de nueva generación ha permitido identificar nuevos genes de varios trastornos mendelianos47. Aportaron por primer vez la prueba de este concepto Ng et al en 2009, empleando secuenciación de exoma completo de 4 sujetos no emparentados que padecían el trastorno muy poco frecuente de síndrome de Freeman-Sheldon y 8 controles, para identificar un único gen candidato MYH3 para este trastorno mendeliano48. En nuevos estudios realizados con exoma completo y genoma completo, se han identificado también nuevos genes candidatos en otros varios trastornos como el síndrome de Kabuki49, la miocardiopatía dilatada autosómica recesiva50 y la enfermedad de Charcot-Marie-Tooth51, lo cual demuestra la utilidad de estas estrategias de secuenciación de nueva generación.
Modificadores genéticos de enfermedades cardiovascularesLos estudios para identificar modificadores genéticos que expliquen la reducida penetrancia y la expresividad variable de estas canalopatías y miocardiopatías potencialmente mortales que a menudo se observan en sujetos no relacionados e incluso dentro de una misma familia están en la vanguardia de la investigación de genética cardiovascular en el futuro previsible.
Estudios de asociación de genoma completoLa finalización de la versión final del proyecto Genoma Humano en 2003 y la aparición de métodos de determinación del genotipo de alto rendimiento han aportado instrumentos para realizar estudios de asociación de genoma completo (GWAS) en poblaciones amplias. Dichos estudios utilizan la secuenciación de miles de individuos para acceder a polimorfismos de nucleótido único (SNP) frecuentes que se asocian a rasgos complejos (no mendelianos o poligénicos). Ya se han realizado varios GWAS que identifican miles de SNP de los que se ha descrito asociación con diversas enfermedades o rasgos complejos incluidos en el Catalog of Published GWAS (www.genoma.gov/26525384)52. Recientemente, se han realizado GWAS para identificar SNP asociados a factores de riesgo cardiovascular que predisponen a enfermedad coronaria53, así como rasgos electrocardiográficos compatibles con un aumento del riesgo de arritmias ventriculares y muerte súbita cardiaca, como intervalo PR, duración del QRS e intervalo QT54–56. Un metaanálisis de estos estudios puso de manifiesto la asociación más intensa existente entre variantes del gen NOS1AP (capón) y la duración del intervalo QT, y los estudios de seguimiento han resaltado la importancia de la vía de la óxido nítrico sintasa en la función miocárdica y la repolarización del potencial de acción57,58. El perfeccionamiento continuo de los GWAS parece prometedor para identificar factores genéticos clave que puedan explicar en parte la reducida penetrancia y la expresividad variable que a menudo se observan en las canalopatías y miocardiopatías cardiacas que se comentan aquí.
Los micro-ARN como posibles factores contribuyentes y modificadores de la enfermedad cardiovascularLos micro-ARN pueden subyacer también a la susceptibilidad a la enfermedad cardiaca y la variación fenotípica. Los micro-ARN son represores postranscripcionales de la expresión génica que se unen a la región 3’ no traducida de los transcriptos del gen diana para eliminar los micro-ARN que no deben expresarse en determinado momento o determinado tipo de célula. Se ha planteado la hipótesis de que este mecanismo contribuye a producir la heterogeneidad observada en la enfermedad individual. Hasta la fecha, varios estudios han puesto de relieve que los polimorfismos del lugar de fijación de micro-ARN pueden causar o modular muchos fenotipos de enfermedad, como el asma59 y la ataxia de Friedrich60, y más recientemente la desregulación de micro-ARN específicos se ha relacionado con el desarrollo y la modulación de enfermedades cardiacas61,62. Por ejemplo, los resultados de estudios en los que se han utilizado chips de expresión de micro-ARN indican distintas regulaciones de micro-ARN en diversas enfermedades (como MCH, miocardiopatía dilatada y miocardiopatía isquémica) en comparación con el corazón normal63. Muy recientemente, Amin et al. han demostrado primorosamente de qué forma los polimorfismos de lugar de fijación de micro-ARN en la región no traducida 3’ de los canales de potasio Kv7.1 codificados por KCNQ1 actúan como posible modificador con especificidad alélica del intervalo QT y los síntomas en los pacientes con LQT1, superando cualquier otro efecto modificador genético64. En su estudio se identificaron tres SNP existentes de manera natural en la región no traducida 3’ de KCNQ1, de tal manera que la presencia del haplotipo alélico menor en el alelo normal acentuaba el fenotipo LQT1 al inhibir los transcriptos de KCNQ1 sanos, mientras que la presencia del haplotipo alélico menor en el alelo mutante atenuaba el fenotipo LQT1 al inhibir los transcriptos de KCNQ1 mutante. Estos resultados no sólo ilustran un nuevo concepto en nuestro conocimiento de los determinantes genéticos modificadores de la enfermedad en los trastornos mendelianos, sino que pueden explicar también una parte importante de la expresividad variable y la reducida penetrancia que se observa en las canalopatías cardiacas65.
Miocardiocitos derivados de células madre pluripotenciales inducidasLos avances recientes en la programación y reprogramación celulares han aportado nuevas vías para conocer la etiología de enfermedades complejas. El descubrimiento innovador —que valió un premio Nobel a los Dres. Takahashi y Yamanaka— de que los fibroblastos de ratón embrionarios y del adulto pueden adquirir características similares a las de las células madre embrionarias tras una transducción retroviral con cuatro factores de transcripción (Oct3/4, Sox2, Klf4 y c-Myc) ha revolucionado el campo de la biología de las células madre66. En 2007, esta tecnología se aplicó a células humanas y actualmente los investigadores pueden generar células madre pluripotenciales inducidas (CMPi) a partir de los fibroblastos derivados de una biopsia cutánea del propio paciente empleando la misma mezcla de factores de transcripción o una mezcla determinada de manera independiente67,68. En 2009, Zhang et al69 presentaron por primera vez la demostración de que las CMPi de origen humano pueden diferenciarse para dar lugar a miocardiocitos funcionales.
Posteriormente se han generado varios modelos de cardiocitos derivados de CMPi que son específicos de canalopatías y miocardiopatías. Por ejemplo, se han publicado al menos cinco líneas de CMPi diferentes procedentes de pacientes con LQTS, y en todos estos estudios los investigadores reprodujeron el fenotipo cardiaco de repolarización prolongada y aumento de la arritmogénesis que se asocia a la enfermedad70–74. En otros estudios se han utilizado CMPi derivadas de pacientes con LQTS para investigar nuevos fármacos para el tratamiento de la enfermedad74,75. El uso de CMPi se ha propuesto también para investigar la prolongación de los potenciales de acción a causa de la posible cardiotoxicidad inducida por fármacos con medicaciones nuevas o ya existentes, con objeto de mejorar la seguridad y el desarrollo de los medicamentos76. Mediante la generación de CMPi procedentes de múltiples familiares afectados que muestran diversas expresiones de la enfermedad, esta tecnología permitirá una caracterización más precisa de las variantes identificadas en los pacientes, así como de su carácter patogénico o benigno, y el examen de los mecanismos que subyacen a la baja penetrancia y la expresividad variable que se dan con frecuencia en los trastornos eléctricos cardiacos. La previsión importante a este respecto es que las CMPi facilitarán el desarrollo de nuevos métodos terapéuticos y el tratamiento personalizado de los pacientes con enfermedades hereditarias.
CONCLUSIONESEn los últimos 20 años, hemos asistido a avances genómicos que nos han permitido profundizar en nuestro conocimiento y nuestra apreciación del papel de la genética en las enfermedades cardiovasculares. A medida que continuemos descubriendo nuevos genes y mecanismos de enfermedades, ciertamente aumentará el número de pruebas genéticas disponibles. Para las cuatro canalopatías/miocardiopatías hereditarias específicas que se detallan en esta revisión, continuamos ampliando en investigación nuestro conocimiento de los mecanismos de enfermedad subyacentes que continuarán dando margen a nuevas traslaciones al ámbito clínico y una interpretación aún mejor de los resultados de las pruebas genéticas, con lo que se mejorará el paso de la investigación al uso clínico. La aplicación de las pruebas genéticas como instrumento diagnóstico/pronóstico en algunas de las enfermedades cardiacas ha impulsado a la comunidad cardiológica a conocer bien el lenguaje de la medicina genómica e interpretar con conocimiento de causa los resultados de las pruebas genéticas. A medida que las nuevas tecnologías, como la secuenciación de exoma/genoma completo y los miocardiocitos generados a partir de CMPi, mejoren y pasen a ser más asequibles, dispondremos de nuevos instrumentos para avanzar hacia nuestro objetivo de una medicina personalizada para tomar decisiones juiciosas con las familias a las que se examina respecto a la presencia o ausencia de uno de estos trastornos de arritmia cardiaca genética potencialmente letales, aunque muy aptos para el tratamiento.
CONFLICTO DE INTERESESEl Dr. Ackerman es consultor de Biotronik, Boston Scientific, Medtronic, St. Jude Medical Inc. y Transgenomic. La propiedad intelectual derivada del programa de investigación de MJA llevó a contratos de licencia en 2004 entre Mayo Clinic Health Solutions (anteriormente Mayo Medical Ventures) y PGxHealth (anteriormente Genaissance Pharmaceuticals, recientemente adquirida por Transgenomic). No hay otros conflictos de intereses.