El diagnóstico de la miocardiopatía arritmogénica del ventrículo derecho (MAVD) puede resultar difícil. La evidencia reciente indica que la evolución natural de esta enfermedad incluye una primera fase oculta, que se caracteriza por exacerbaciones agudas de la inflamación miocárdica y arritmias ventriculares con peligro para la vida, que se producen antes de la aparición de las manifestaciones clínicas clásicas y contribuyen a su patogenia y su progresión1. Prueba de ello son las descripciones de casos de MAVD que se presentan en forma de episodios recurrentes de miocarditis en pacientes jóvenes con signos de inflamación miocárdica en la cardiorresonancia magnética2. En vez de la clásica sustitución de miocitos del miocardio del ventrículo derecho (VD) por tejido fibroso o fibroadiposo3, a menudo se pueden observar infiltrados inflamatorios en las áreas afectadas4. Este artículo tiene como finalidad ilustrar esta asociación y aportar un argumento convincente respecto a la conveniencia de realizar un estudio detallado del VD en los pacientes jóvenes que presentan arritmias ventriculares y signos de miocarditis activa o antecedentes de ella.
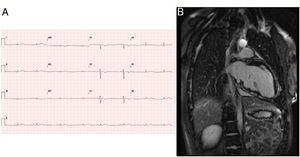
La paciente 1, una niña anteriormente sana, fue atendida a la edad de 15 años por una muerte súbita cardiaca abortada mientras practicaba un deporte de competición. El ritmo era de una taquicardia ventricular sin pulso. En el electrocardiograma (ECG) inicial se observó bajo voltaje y una inversión de la onda T en las derivaciones derechas (figura 1), que se consideraron normales para su edad. El padre tenía el mismo patrón de onda T, pero por lo demás no había antecedentes familiares de interés. Una semana antes del episodio actual, se le diagnosticó una traqueobronquitis, con 1 día de fiebre. Durante el ingreso actual, la evolución fue buena. Un registro Holter de 24 h mostró la presencia de extrasístoles ventriculares polimórficas (28 extrasístoles/h). La cardiorresonancia magnética reveló signos indirectos de inflamación activa con una señal SSFP miocárdica del ventrículo izquierdo (VI) espontánea aumentada, y múltiples localizaciones de realce tardío de gadolinio (RTG), que eran más evidentes en la pared inferior del VI (figura 1). La fracción de eyección del VI (FEVI) fue del 58%, y el volumen telediastólico era normal (85 ml/m2). La fracción de eyección del VD era del 48%, y el volumen telediastólico se encontraba en el límite superior de la normalidad (108ml/m2). Se apreció una discinesia leve del tracto de salida del VD (TSVD) (), pero no hubo consenso al respecto. En ese momento, se estableció un diagnóstico de presunción de miocarditis aguda, a pesar del estudio etiológico negativo. Se implantó un desfibrilador automático implantable (DAI). En el examen genético mediante método de secuenciación para la detección sistemática de variantes comunes o raras, se observó una variante previamente desconocida en forma heterocigota: c.1840delC en el gen PKP2, causante de una truncación de la proteína, que es diagnóstica de la MAVD. El padre es portador del gen, y muestra solo anomalías del ECG. No se estudió a familiares más lejanos. La paciente ha recibido seguimiento y se ha mantenido bien durante 2 años.

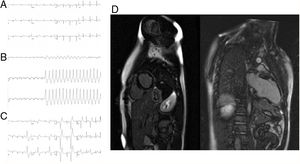
El paciente 2, un niño de 13 años, fue remitido a causa de una extrasistolia ventricular con morfología de bloqueo de rama izquierda del haz de His en el último de los ECG realizados cada 2 años para la práctica de un deporte de competición. Los ECG mostraron una inversión de la onda T en las derivaciones precordiales derechas (figura 2), que se consideró normal para su edad. Los antecedentes familiares fueron negativos y el paciente había estado sano anteriormente, excepto por un episodio de dolor torácico opresivo 2 años antes, que duró unos pocos días y precedió a la aparición de la extrasistolia. En la evaluación actual el paciente mencionó una sensación de aturdimiento y palpitaciones durante la práctica de deportes. La ecocardiografía mostró una leve dilatación del VD. En la prueba de esfuerzo se provocó una taquicardia ventricular del TSVD no sostenida (figura 2) y múltiples extrasístoles del TSVD aisladas (figura 2). La cardiorresonancia magnética mostró una dimensión y una función del VI normales, pero con un amplio RTG subepicárdico (figura 2). Había una dilatación leve del VD (115ml/m2), y la fraccióon de eyección era del 39%, con una discinesia leve en el ápex y el TSVD (). Dados los antecedentes de dolor torácico del paciente, el patrón de RTG y la discinesia del VD, surgieron dudas acerca del diagnóstico correcto: tejido cicatrizal derivado de un episodio previo de miocarditis o MAVD expresada por una exacerbación inflamatoria previa. Se instauró tratamiento con un bloqueador beta. Mientras estaba a la espera del implante de un desfibrilador automático, el paciente presentó un episodio de taquicardia del TSVD sostenida. En el estudio electrofisiológico de seguimiento se indujo esta taquicardia y se realizó con éxito la ablación. Se indujo otra forma de taquicardia ventricular más rápida originada en el ápex, pero que no se mantuvo lo suficiente para permitir un mapeo. Se implantó un DAI subcutáneo. En el ECG de promediación de señal de alta resolución, se observaron potenciales tardíos. Las pruebas genéticas realizadas con método de secuenciación mostraron una variante no descrita con anterioridad en forma heterocigota: c.224-2T>C en el gen PKP2, causante de una truncación de la proteína y diagnóstica de una MAVD. Los familiares de primer grado rechazaron someterse al estudio. El paciente ha sido tratado con sotalol y en el seguimiento de 1 año ha estado asintomático.

Paciente 2. A: ECG inicial con inversión de la onda T en las derivaciones precordiales derechas. B: taquicardia ventricular durante la prueba de esfuerzo, con eje inferior. C: extrasístoles ventriculares de origen en el TSVD. D: cardiorresonancia magnética que muestra un realce tardío de gadolinio subepicárdico amplio, sobre todo en la pared inferolateral del ventrículo izquierdo.
Estos casos ilustran cómo la MAVD puede manifestarse en una etapa temprana de la vida por arritmias ventriculares potencialmente mortales y un RTG subepicárdico inferolateral. Este es un patrón de RTG frecuente tanto en la miocarditis5 como en la MAVD con afección del VI1. En consecuencia, la miocarditis, que tiene mayor prevalencia en estos individuos jóvenes, puede diagnosticarse rápidamente. Una consideración detallada de otras pistas diagnósticas, como el tamaño y la contractilidad regional del VD de nuestros pacientes, puede llevar a la realización de pruebas genéticas completas6 y a la identificación de la enfermedad subyacente. De hecho, ambos pacientes cumplen los criterios del grupo de trabajo3 (3 criterios mayores + 1 criterio menor y 2 criterios mayores + 2 criterios menores respectivamente). El establecimiento de un diagnóstico correcto de MAVD tiene importantes consecuencias para el futuro tratamiento y la estratificación del riesgo familiar.