El tratamiento quimioterápico de los pacientes con neoplasias malignas es potencialmente cardiotóxico, y la disfunción ventricular secundaria a cardiotóxicos (DV-CTOX) es una de las complicaciones más frecuentes1. Su presentación depende del riesgo basal del paciente, por lo que es fundamental una adecuada estratificación. Actualmente se recomienda considerar determinadas características clínicas (edad, factores de riesgo cardiovascular, enfermedad cardiaca), la asociación con otros tratamientos cardiotóxicos y, en el caso de las antraciclinas, la dosis acumulada2. Sin embargo, hay casos que desarrollan una disfunción ventricular desproporcionada en comparación con el riesgo basal estimado, lo que indica una susceptibilidad individual a la enfermedad. En este sentido, se han descrito familias que comparten casos de miocardiopatía dilatada y DV-CTOX, lo que apunta a una base genética común3. Recientemente se ha demostrado que las variantes de tipo truncamiento en el gen TTN son significativamente prevalentes en pacientes con DV-CTOX y se relacionan con mayor incidencia de eventos adversos4. La información sobre otros genes potencialmente asociados es escasa y no hay recomendaciones específicas sobre la realización de estudio genético en este contexto.
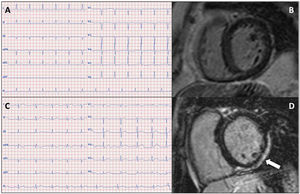
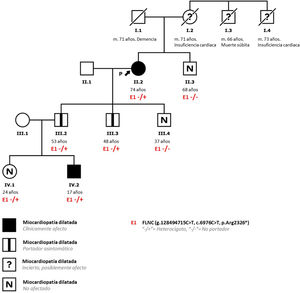
Se presenta el caso de una mujer de 74 años hipertensa, bien controlada, con antecedente de cáncer de mama tratado con antraciclinas y radioterapia y ecocardiografía previa al tratamiento normal. Pasados 10 años desde el tratamiento, la paciente desarrolló clínica de insuficiencia cardiaca y se detectó miocardiopatía dilatada con disfunción sistólica grave del ventrículo izquierdo (VI). El electrocardiograma mostraba escasa R en V2-V3 y ondas T aplanadas en cara inferior y V6 y negativas en V4-V5 (figura 1A). Se realizó una coronariografía, que mostró arterias coronarias sanas, y una resonancia magnética cardiaca, en la que no se observó realce tardío de gadolinio (figura 1B). En la historia familiar destacaba la presencia de enfermedad cardiovascular relativamente precoz por vía materna, sin detectarse cardiopatía por ecocardiografía en 1 hermano y 3 hijos varones (figura 2). Para completar la evaluación, se realizó el estudio genético mediante secuenciación de nueva generación, con panel de miocardiopatía dilatada (121 genes), que identificó una variante patogénica en el gen FLNC (p.Arg2326*) descrita previamente en 6 familias, en las que la mayoría de los miembros portadores de la variante presentaban fenotipo de miocardiopatía dilatada y elevada incidencia de arritmias ventriculares.
. B: resonancia magnética cardiaca de la misma paciente, sin realce tardío de gadolinio. C: electrocardiograma del nieto de la paciente caso índice (IV.2). D: resonancia magnética cardiaca del mismo paciente; patrón de realce tardío de gadolinio subepicárdico en las caras inferior y lateral del ventrículo izquierdo (flecha).")
A: electrocardiograma del caso índice (II.2). B: resonancia magnética cardiaca de la misma paciente, sin realce tardío de gadolinio. C: electrocardiograma del nieto de la paciente caso índice (IV.2). D: resonancia magnética cardiaca del mismo paciente; patrón de realce tardío de gadolinio subepicárdico en las caras inferior y lateral del ventrículo izquierdo (flecha).

En el estudio familiar se detectó a 2 hijos portadores asintomáticos con fracción de eyección conservada y sin dilatación, ambos con realce tardío de gadolinio subepicárdico en la pared lateral del VI, ondas T negativas en III y aVF en el electrocardiograma y ausencia de arritmias en registro Holter. Uno de ellos tiene 1 hijo varón de 17 años, federado en fútbol y portador de la variante descrita, que se encuentra asintomático aunque presenta dilatación y disfunción ligera del VI, desviación del eje a la derecha en el electrocardiograma (figura 1C) y patrón de realce tardío de gadolinio subepicárdico en las caras inferior y lateral del VI (figura 1D), sin arritmias en el registro Holter. Su hermana de 24 años es portadora, aunque de momento no presenta fenotipo. En ambos casos se ha recomendado evitar la práctica de deporte de alta intensidad y realizar seguimiento clínico estrecho. Los demás familiares evaluados no son portadores de la variante.
Las variantes de tipo truncamiento en FLNC se han asociado con un fenotipo consistente en miocardiopatía dilatada/arritmogénica de predominio izquierdo, caracterizada por una alta incidencia de arritmias ventriculares y una considerable incidencia de muerte súbita, especialmente a partir de los 40 años de edad, incluso en ausencia de disfunción sistólica grave. Los varones desarrollan el fenotipo más pronto y el pronóstico, en relación con el desarrollo de arritmias y muerte súbita, es peor que para las mujeres5. Para los portadores, se recomienda una resonancia magnética cardiaca para detectar fibrosis miocárdica y un electrocardiograma Holter para detectar arritmias aunque la ecocardiografía sea normal. Se desaconseja en estos casos la práctica de deporte competitivo. A diferencia de lo descrito en la literatura, en esta familia la penetrancia es incompleta y el fenotipo, mayoritariamente benigno. Es posible que el fenotipo de algunos truncamientos en FLNC sea más variable de lo descrito previamente y que desencadenantes adicionales de tipo ambiental influyan de manera importante en su gravedad, como la quimioterapia o la hipertensión arterial en el caso índice o el deporte competitivo en su nieto, que ha favorecido una aparición temprana de la enfermedad.
En los pacientes con cáncer que reciben quimioterapia, la estratificación del riesgo de desarrollar disfunción ventricular secundaria al tratamiento sigue siendo un reto. Actualmente se basa en marcadores clínicos que no son capaces de predecir la susceptibilidad individual. El estudio genético puede ser útil para identificar a los pacientes con una mayor predisposición a sufrir DV-CTOX, especialmente cuando hay antecedentes familiares sugestivos. Además, permite la identificación precoz de familiares en riesgo que pueden beneficiarse de estrategias preventivas orientadas a retrasar la progresión de la enfermedad y reducir las complicaciones.
Los autores confirman que se ha obtenido el consentimiento informado de la paciente y sus familiares para publicar el caso, incluidas las imágenes.
FINANCIACIÓNNo se ha requerido.
CONTRIBUCIÓN DE LOS AUTORESM.L. Peña Peña y M.R. Caballero Valderrama fueron las responsables del caso presentado, realizaron la revisión bibliográfica y la redacción del manuscrito. S. Navarro Herrero y M.P. Serrano Gotarredona informaron las imágenes de resonancia cardiaca y revisaron el manuscrito. J.E. López Haldón contribuyó al enfoque del manuscrito y realizó una revisión crítica de este.
CONFLICTO DE INTERESESNo hay conflicto de intereses.