La miocardiopatía hipertrófica (MCH) se diagnostica por la presencia de una hipertrofia ventricular ≥ 15 mm en ausencia de otras condiciones de sobrecarga que puedan originarla o, en el caso de un familiar de un paciente con MCH, cuando presenta un grosor ≥ 13 mm. A la histología, la desorganización miofibrilar es característica de la enfermedad. Sin embargo, se han descrito casos de MCH asociados a muerte súbita (MS), en los que solo se ha identificado esa desorganización o la presencia de mutaciones en el gen TNNT21. Se presenta a una familia con varias muertes súbitas, para cuyo diagnóstico ha sido clave el estudio anatomopatológico y genético.
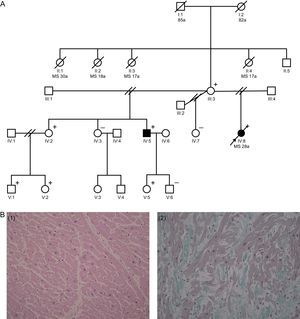
El caso índice es una mujer de 28 años, sin antecedentes personales patológicos, que sufrió MS al entrar en su coche. La autopsia mostró un corazón de 295 g de peso, con un tabique interventricular de 13 mm. En el análisis microscópico se apreció hipertrofia microscópica de fibras y focos aislados de desorden fibrilar (figura B). Entre los antecedentes familiares, destacan 4 hermanas de la madre de la paciente que fallecieron por MS a los 17, 18 y 30 años (figura A). La madre de la paciente tenía 55 años, un ecocardiograma normal y electrocardiograma (ECG) con alteraciones de la repolarización en la cara inferolateral. El caso índice tenía 3 hermanas y 1 hermano de diferentes padres. Una de las hermanas, de 30 años, tenía un ecocardiograma y una cardiorresonancia magnética (CRM) totalmente normales, pero el ECG mostraba descenso del segmento ST en las caras inferior y lateral. Se realizó ecocardiografía de estrés y coronariografía, que no evidenciaron alteraciones. Las otras 2 hermanas presentaban ecocardiogramas y ECG normales. En el hermano se observa ligera hipertrofia del ventrículo izquierdo, con un septo interventricular de 16 mm, y el ECG era similar a los de la hermana viva y la madre (tabla).
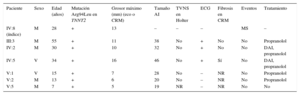
. B: imágenes del estudio histológico de los distintos cortes realizados al tabique y al ventrículo izquierdo de la paciente índice (IV:8); revela la presencia de un miocardio congestivo, integrado por miocitos hipertróficos que se disponen de forma ordenada (panel izquierdo). Se aprecian algunas áreas de pequeño tamaño en la pared libre ventricular izquierda donde dichas fibras se disponen de manera desordenada, alineándose oblicuamente y configurando remolinos (disarray) (panel derecho). a: años; MS: muerte súbita.")
A: árbol genealógico de la familia (cuadrado: varón; círculo: mujer; cuadrado o círculo con relleno: varón o mujer afectado/a fenotípicamente; cuadrado o círculo sin relleno: varón o mujer sano/a; línea diagonal: fallecido; líneas diagonales paralelas: divorcio). B: imágenes del estudio histológico de los distintos cortes realizados al tabique y al ventrículo izquierdo de la paciente índice (IV:8); revela la presencia de un miocardio congestivo, integrado por miocitos hipertróficos que se disponen de forma ordenada (panel izquierdo). Se aprecian algunas áreas de pequeño tamaño en la pared libre ventricular izquierda donde dichas fibras se disponen de manera desordenada, alineándose oblicuamente y configurando remolinos (disarray) (panel derecho). a: años; MS: muerte súbita.
Características clínicas de los portadores de la mutación Arg94Leu en TNNT2
| Paciente | Sexo | Edad (años) | Mutación Arg94Leu en TNNT2 | Grosor máximo (mm) (eco o CRM) | Tamaño AI | TVNS en Holter | ECG | Fibrosis en CRM | Eventos | Tratamiento |
|---|---|---|---|---|---|---|---|---|---|---|
| IV:8 (índice) | M | 28 | + | 13 | – | – | – | MS | – | |
| III:3 | M | 55 | + | 11 | 38 | No | + | No | No | Propranolol |
| IV:2 | M | 30 | + | 10 | 32 | No | + | No | No | DAI, propranolol |
| IV:5 | V | 34 | + | 16 | 46 | No | + | Sí | No | DAI, propranolol |
| V:1 | V | 15 | + | 7 | 28 | No | – | NR | No | Propranolol |
| V:2 | M | 13 | + | 6 | 20 | No | – | NR | No | Propranolol |
| V:5 | M | 7 | + | 5 | 19 | NR | – | NR | No | No |
AI: aurícula izquierda; CRM: cardiorresonancia magnética; DAI: desfibrilador automático implantable; eco: ecocardiograma; ECG: electrocardiograma con alteraciones de la repolarización en cara inferolateral; M: mujer; NR: no realizada; TVNS: taquicardia ventricular no sostenida; V: varón.
Ante la sospecha de MCH familiar con expresión ligera y alta incidencia de MS, se decidió realizar un estudio genético en una muestra de sangre congelada de la paciente fallecida. Se utilizó un panel que estudia mediante next generation sequencing varios genes relacionados con miocardiopatías y canalopatías, por la posibilidad de que hubiese alguna canalopatía subyacente no diagnosticada en las pacientes fallecidas. Se identificó la mutación Arg94Leu en troponina T (TNNT2). Esta mutación se describió inicialmente en una familia británica con alta prevalencia de MS precoz (< 45 años) y diagnóstico compatible con MCH. Estos pacientes no tenían antecedentes de enfermedad, por lo que la MS fue la primera manifestación clínica. Los hallazgos de la necropsia consistieron en la ausencia de hipertrofia macroscópica, aunque el análisis histológico mostró fibrosis difusa y desorganización de miocitos. De hecho, este trabajo fue uno de los primeros en establecer que las mutaciones en TNNT2 podían asociarse a MS aun en ausencia de hipertrofia manifiesta2,3. Se han descrito otras 2 mutaciones que afectan al mismo residuo (Arg94Cys, Arg94His), por lo que este parece ser un punto proclive a las mutaciones. Esto indicaría que cualquier cambio en la secuencia de aminoácidos en este punto sería mal tolerado. La información clínica disponible sobre los portadores de dichas mutaciones coincide, por ejemplo, con lo observado en la mutación Arg92Gln de varias familias mallorquinas, recientemente publicado4. Algunos trabajos que han evaluado estudios microscópicos en portadores de mutaciones en troponina T, indican que estas causan menos hipertrofia y fibrosis que otras mutaciones sarcoméricas, pero más desorden. Este puede ser el sustrato que explique el elevado riesgo arrítmico5.
El estudio genético del resto de la familia mostró que la madre, una hermana (y sus 2 hijos) y el hermano (y una de sus hijas) eran portadores de la mutación identificada. Aunque las últimas guías no recomiendan inicialmente el implante de desfibrilador automático implantable únicamente por la presencia de una mutación determinada, por preferencia de los pacientes (que no tenían ningún factor de riesgo, salvo la MS familiar) (tabla), se implantó como prevención primaria desfibrilador a sus 2 hermanos de 33 y 35 años. A los demás portadores, todos menores de 16 años excepto la madre del caso índice (55 años), se les hace un seguimiento estrecho en consulta.
En conclusión, los antecedentes familiares, el estudio anatomopatológico y el estudio genético son clave en el estudio de la MS6. Los datos aportados por la familia presentada indican que la MCH debida a la mutación Arg94Leu en TNNT2 puede presentarse sin hipertrofia macroscópica, con alteraciones en ECG y desorden en el estudio histológico. El riesgo de MS en portadores es alto, por lo que debe realizarse una estricta estratificación del riesgo.