
El síndrome de QT largo, el síndrome de QT corto, el síndrome de Brugada y la taquicardia ventricular polimórfica catecolaminérgica son trastornos eléctricos primarios que predisponen a muerte súbita en ausencia de cardiopatía estructural. La estratificación del riesgo en estas enfermedades es fundamental para identificar a los pacientes con peor pronóstico. En el síndrome de QT largo y el síndrome de Brugada, numerosos estudios han establecido que los principales factores pronósticos son clínicos y se basan en la presencia de síntomas previos, los hallazgos del ECG, el sexo y, en el caso del síndrome de Brugada, la inducibilidad de arritmias en el estudio electrofisiológico. Por el escaso número de casos descritos, actualmente disponemos de poca información acerca de las variables implicadas en el pronóstico del síndrome de QT corto y la taquicardia ventricular polimórfica catecolaminérgica. Esta revisión resume nuestro conocimiento actual sobre la estratificación del riesgo en estos cuatro trastornos hereditarios arritmogénicos.
Palabras clave
La muerte súbita (MS) es la complicación más temida de toda cardiopatía. Un escenario especialmente dramático es el de la MS cardiaca que ocurre en sujetos jóvenes o sin enfermedad previa conocida, en cuyas autopsias puede revelarse un corazón estructuralmente normal en hasta el 30% de los casos, según las series1. La MS cardiaca en ausencia de cardiopatía estructural suele responder a un trastorno arritmogénico de origen genético. Son síndromes relacionados con MS cardiaca el síndrome de QT largo (SQTL), el síndrome de Brugada (SBr), el síndrome de QT corto (SQTC) y la taquicardia ventricular polimórfica catecolaminérgica (TVPC). Característicamente, estas enfermedades tienen una penetrancia variable, de forma que no todos los portadores genéticos manifiestan el fenotipo de la enfermedad, y entre los que lo tienen, el grado de presentación clínica es extremadamente variable, desde la ausencia completa de síntomas hasta la MS en edad joven. Ello conlleva que, ante el diagnóstico de cualquiera de estas entidades, una de las prioridades del médico tratante sea identificar a los pacientes con mayor riesgo de MS. Este artículo se centra precisamente en las herramientas pronósticas que han demostrado utilidad en la estratificación del riesgo de MS para cada una de estas enfermedades.
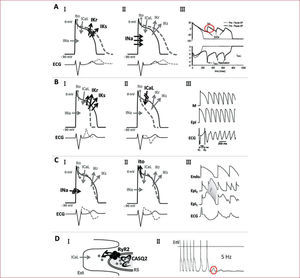
Síndrome de qt largoFisiopatología y diagnósticoEl SQTL es un trastorno hereditario caracterizado por la presencia de un intervalo QT alargado en el ECG y la propensión a las arritmias ventriculares polimórficas, típicamente torsade de pointes. Su preva-lencia se estima en torno a 1:3.000–1:5.000 individuos, aunque estudios recientes sobre genotipificación indican que probablemente el número de sujetos afectados sea mucho mayor2. Hasta la fecha, se han descrito diversas formas de SQTL, cada una de ellas relacionada con la mutación de un gen diferente. Los SQTL tipos 1, 2 y 3 son los más frecuentes en la práctica clínica y representan en conjunto más del 90% de los casos de SQTL3. Los SQTL tipos 1 y 2 se producen por mutaciones en los genes KCNQ1 y KCNH2, que producen una pérdida de función de los canales de potasio de activación lenta (IKs) y rápida (IKr), respectivamente. Por su parte, el SQTL tipo 3 responde a mutaciones en el gen SCN5A, que codifica la subunidad alfa del canal de sodio cardiaco y cuyo efecto es la hiperfunción de este2. Los demás SQTL (tipos 4 a 12) son anecdóticos en la práctica clínica. El síndrome de Jervell y Lange-Nielsen, la primera forma de SQTL descrita hace más de 50 años y asociada a sordera, es la forma recesiva constituida por mutaciones homocigóticas o compuestas de los SQTL tipos 1 y 54. Sea por un mecanismo u otro, la característica común de todos los tipos de SQTL es una mayor duración del potencial de acción celular, lo cual prolonga la repolarización y explica el intervalo QT alargado que se observa en el ECG (figs. 1A I y II). En estas circunstancias se ve favorecida la aparición de pospotenciales precoces, fundamentalmente por reactivación del canal de calcio ICaL, los cuales pueden actuar como desencadenantes de arritmias ventriculares y torsade de pointes (fig. 1A III)5.
 tipos 1 y 2, producidos por pérdida de función de las corrientes de potasio (I), y SQTL tipo 3, producido por hiperfunción del canal de sodio (II); ambas situaciones favorecen la aparición de pospotenciales precoces (III), el desencadenante de arritmias ventriculares en estos pacientes (III reproducido con autorización de Rudy et al5). B: síndrome de QT corto (SQTC) tipos 1, 2 y 3, producidos por aumento de las corrientes de potasio (I), y SQTC tipos 4 y 5, producidos por hipofunción del canal de corrientes de calcio ICaL (II), en este caso asociados a fenotipo de síndrome de Brugada; el acortamiento del periodo refractario, junto con el aumento de dispersión de la repolarización, favorece la aparición de arritmias auriculares y ventriculares (III) (III reproducido con autorización de Patel et al31). C: síndrome de Brugada (SBr), producido por hipofunción del canal de sodio (I) y, como se ha descrito recientemente, por el aumento indirecto de las corrientes Ito (II); el desequilibrio de corrientes positivas de entrada y salida de la célula en la fase 1 del potencial de acción produce dispersión de la repolarización epicárdica y transmural, lo cual favorece arritmias ventriculares por un mecanismo de reentrada en fase 2 (III, reproducido con autorización de Benito et al38). D: taquicardia ventricular polimórfica catecolaminérgica (TVPC), producida por mutaciones en los genes RyR2 (TVPC tipo 1) o CASQ2 (TVPC tipo 2), que producen liberación excesiva de Ca procedente del retículo sarcoplásmico (RS) (I), lo cual favorece la aparición de pospotenciales tardíos, que a su vez pueden desencadenar la arritmia (II, reproducido con autorización de Liu et al51).")
Esquema fisiopatológico de las principales enfermedades arritmogénicas hereditarias sin cardiopatía estructural asociada. A: síndrome de QT largo (SQTL) tipos 1 y 2, producidos por pérdida de función de las corrientes de potasio (I), y SQTL tipo 3, producido por hiperfunción del canal de sodio (II); ambas situaciones favorecen la aparición de pospotenciales precoces (III), el desencadenante de arritmias ventriculares en estos pacientes (III reproducido con autorización de Rudy et al5). B: síndrome de QT corto (SQTC) tipos 1, 2 y 3, producidos por aumento de las corrientes de potasio (I), y SQTC tipos 4 y 5, producidos por hipofunción del canal de corrientes de calcio ICaL (II), en este caso asociados a fenotipo de síndrome de Brugada; el acortamiento del periodo refractario, junto con el aumento de dispersión de la repolarización, favorece la aparición de arritmias auriculares y ventriculares (III) (III reproducido con autorización de Patel et al31). C: síndrome de Brugada (SBr), producido por hipofunción del canal de sodio (I) y, como se ha descrito recientemente, por el aumento indirecto de las corrientes Ito (II); el desequilibrio de corrientes positivas de entrada y salida de la célula en la fase 1 del potencial de acción produce dispersión de la repolarización epicárdica y transmural, lo cual favorece arritmias ventriculares por un mecanismo de reentrada en fase 2 (III, reproducido con autorización de Benito et al38). D: taquicardia ventricular polimórfica catecolaminérgica (TVPC), producida por mutaciones en los genes RyR2 (TVPC tipo 1) o CASQ2 (TVPC tipo 2), que producen liberación excesiva de Ca procedente del retículo sarcoplásmico (RS) (I), lo cual favorece la aparición de pospotenciales tardíos, que a su vez pueden desencadenar la arritmia (II, reproducido con autorización de Liu et al51).
Hoy, la identificación de la mutación causal mediante estudio genético permite confirmar el diagnóstico de SQTL hasta en el 72% de los casos2. Sin embargo, en la práctica clínica el ECG sigue siendo la herramienta más útil en el diagnóstico, ya que permite identificar un intervalo QT alargado en la mayoría de los pacientes. Se consideran patológicos los intervalos QT corregidos para la frecuencia cardiaca (QTc, fórmula de Bazett: QTc=QT /RR0,5) que superan los 460 ms en niños menores de 15 años, los 450 ms en varones adultos y los 470 ms en mujeres adultas2. Además, la morfología del intervalo QT y la onda T puede ser útil para diagnosticar algunos casos con QTc borderline y al mismo tiempo puede aportar información orientativa sobre el genotipo (fig. 2A)6. Basándose en los datos del ECG, la presencia de síntomas y la historia familiar, Schwartz et al7 propusieron en 1993 una escala con tres grados de probabilidad (alta, intermedia y baja), útil para el diagnóstico clínico. Sin embargo, aunque de alta especificidad, la escala carece de suficiente sensibilidad para ser aplicable en ciertos contextos clínicos, como en el cribado familiar, para el que actualmente se recomienda realizar directamente estudio genético8.
 tipo 3, con ondas T estrechas tras un segmento ST largo; tipo 2, con ondas T de bajo voltaje y a menudo con muesca, y tipo 1, con ondas T de base ancha (reproducido con autorización de Moss et al6). B: síndrome de QR corto (SQTC) tipo 1, con ondas T estrechas y picudas (adaptado de Brugada et el28); tipo 2, con ondas menos picudas (adaptado de Bellocq et al29), y tipo 5, intervalo QT corto combinado con signo de Brugada tras administración de ajmalina (reproducido con autorización de Antzelevitch et al30). C: derivaciones precordiales derechas en el síndrome de Brugada, que muestran el signo típico y el único diagnóstico del síndrome: elevación descendente del segmento ST seguido de T negativas. D: ECG normal en reposo y aparición de taquicardia bidireccional con el ejercicio, característico de taquicardia ventricular polimórfica catecolaminérgica (TVPC) (reproducido con autorización de Liu et al51).")
Características electrocardiográficas en los cuatro trastornos hereditarios arritmogénicos. A: síndrome de QT largo (SQTL) tipo 3, con ondas T estrechas tras un segmento ST largo; tipo 2, con ondas T de bajo voltaje y a menudo con muesca, y tipo 1, con ondas T de base ancha (reproducido con autorización de Moss et al6). B: síndrome de QR corto (SQTC) tipo 1, con ondas T estrechas y picudas (adaptado de Brugada et el28); tipo 2, con ondas menos picudas (adaptado de Bellocq et al29), y tipo 5, intervalo QT corto combinado con signo de Brugada tras administración de ajmalina (reproducido con autorización de Antzelevitch et al30). C: derivaciones precordiales derechas en el síndrome de Brugada, que muestran el signo típico y el único diagnóstico del síndrome: elevación descendente del segmento ST seguido de T negativas. D: ECG normal en reposo y aparición de taquicardia bidireccional con el ejercicio, característico de taquicardia ventricular polimórfica catecolaminérgica (TVPC) (reproducido con autorización de Liu et al51).
La mayoría de los pacientes con SQTL permanecen asintomáticos, pero a pesar de ello una proporción importante sufre eventos a lo largo de su vida. Priori et al3 analizaron la historia natural de 647 pacientes con diagnóstico genético confirmado de SQTL tipos 1, 2 y 3, y observaron, en el conjunto de pacientes, una incidencia acumulada de MS o parada cardiaca del 13% antes de los 40 años y antes de haber iniciado cualquier tratamiento, porcentaje que ascendía hasta el 36% al incluir también síncope como evento cardiaco. Característicamente, los eventos en los pacientes con SQTL aparecen ante determinadas situaciones desencadenantes, en las que de una u otra forma se hace más manifiesto el defecto del canal mutado9. Así, los pacientes con SQTL tipo 1 presentan síntomas típicamente en situaciones de descarga adrenérgica, como en el ejercicio, al hacerse evidente la pobre adaptación a la taquicardia por defecto de IKs. En cambio, los síntomas en el SQTL tipo 3 aparecen en contextos de bradicardia (típicamente durante el reposo nocturno), situación en la que prevalece el defecto del canal de sodio, de recuperación lenta. Los desencadenantes de los síntomas en el SQTL tipo 2 son más variados, pero habi-tualmente se relacionan con situaciones de emociones intensas e inesperadas o con el reposo9.
Diversos factores pueden modular el curso de la enfermedad. En los últimos años, gran parte de la investigación en SQTL se ha centrado precisamente en la identificación de estas variables y en el estudio de su significado pronóstico. En este sentido, ha sido fundamental la información obtenida del Registro Internacional de SQTL, que actualmente incluye más de 1.200 casos índice más sus familiares2.
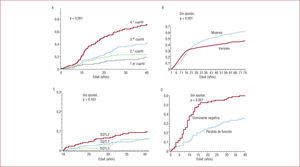
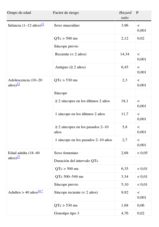
La duración del intervalo QTc se confirmó como factor pronóstico ya en los primeros estudios de SQTL10. Priori et al3 comprobaron, además, que el efecto del intervalo QTc es continuo, de forma que los pacientes con intervalos QTc en el tercer cuartil tienen peor pronóstico que los pacientes en el primer cuartil (hazard ratio [HR]=5,34; intervalo de confianza [IC] del 95%, 2,82–10,13), y los pacientes con intervalos QTc en el cuarto cuartil tienen aún peor pronóstico (HR=8,36; IC del 95%, 2,53–27,21) (fig. 3A). Es importante destacar que ello se confirmó sólo para los pacientes con SQTL tipos 1 y 2, y no para los pacientes con SQTL tipo 3. Estudios recientes obtenidos del Registro Internacional de SQTL, realizados en diferentes franjas de edad y con objeto de analizar el riesgo sólo de MS o parada cardiaca (excluyendo síncope), establecen que un intervalo QTc > 500 ms incrementa el riesgo de MS o parada cardiaca entre 2 y 3 veces durante la infancia y la edad adulta, mientras que el mismo riesgo se ve a partir de valores > 530 ms en la adolescencia y más allá de los 40 años de edad (tabla 1)11–14. Para la estratificación del riesgo, debe tomarse como valor de QTc el más largo observado en el mismo paciente durante todo su seguimiento15. Ello obliga a valorar repetidamente a los pacientes con SQTL.
, el sexo (B, reproducido con autorización de Goldenberg et al2), el genotipo (C, reproducido con autorización de Sauer et al13) y el tipo de mutación en KCNQ1 (D, reproducido con autorización de Moss et al18) en el pronóstico de los pacientes con SQTL. Nótese que en C se representan únicamente los eventos mayores, MS o parada cardiaca. MS: muerte súbita; SQTL: síndrome de QT largo.")
Efecto de la duración del intervalo QTc (A, adaptado con autorización de Priori et al44), el sexo (B, reproducido con autorización de Goldenberg et al2), el genotipo (C, reproducido con autorización de Sauer et al13) y el tipo de mutación en KCNQ1 (D, reproducido con autorización de Moss et al18) en el pronóstico de los pacientes con SQTL. Nótese que en C se representan únicamente los eventos mayores, MS o parada cardiaca. MS: muerte súbita; SQTL: síndrome de QT largo.
Factores de riesgo de muerte súbita o parada cardiaca en pacientes con síndrome de QT largo en los diferentes grupos de edad.
| Grupo de edad | Factor de riesgo | Hazard ratio | P |
| Infancia (1–12 años)11 | Sexo masculino | 3,96 | < 0,001 |
| QTc > 500 ms | 2,12 | 0,02 | |
| Síncope previo | |||
| Reciente (< 2 años) | 14,34 | < 0,001 | |
| Antiguo (≥ 2 años) | 6,45 | < 0,001 | |
| Adolescencia (10–20 años)12 | QTc > 530 ms | 2,3 | < 0,001 |
| Síncope | |||
| ≥ 2 síncopes en los últimos 2 años | 18,1 | < 0,001 | |
| 1 síncope en los últimos 2 años | 11,7 | < 0,001 | |
| ≥ 2 síncopes en los pasados 2–10 años | 5,8 | < 0,001 | |
| 1 síncope en los pasados 2–10 años | 2,7 | < 0,001 | |
| Edad adulta (18–40 años)13 | Sexo femenino | 2,68 | < 0,05 |
| Duración del intervalo QTc | |||
| QTc > 500 ms | 6,35 | < 0,01 | |
| QTc 500–549 ms | 3,34 | < 0,01 | |
| Síncope previo | 5,10 | < 0,01 | |
| Adultos > 40 años14,* | Síncope reciente (< 2 años) | 9,92 | < 0,001 |
| QTc > 530 ms | 1,68 | 0,06 | |
| Genotipo tipo 3 | 4,76 | 0,02 |
En el conjunto de pacientes con SQTL, el sexo como variable aislada no marca significativamente el pronóstico3. Sin embargo, estudios recientes reflejan que existen diferencias en el comportamiento del SQTL entre varones y mujeres cuando se analizan por separado diferentes grupos de edad (fig. 3B y tabla 1). Así, durante la infancia, el riesgo de síncope, MS o parada cardiaca es mayor en niños que en niñas, especialmente en los portadores de un SQTL tipo 12. Cuando el análisis se restringe sólo a la documentación de MS o parada cardiaca, la diferencia entre niños y niñas persiste (el 5 frente al 1% de eventos respectivamente entre 1 y 12 años; p < 0,001) y se cumple por igual para todos los genotipos11. Durante la adolescencia, el riesgo entre varones y mujeres se iguala12, y a partir de aproximadamente los 20 años se invierte, de forma que las mujeres incrementan en 2,68 el riesgo de MS o parada cardiaca respecto a los varones de la misma franja de edad13. Se piensa que esta variabilidad responde en parte a influencias hormonales. De hecho, un estudio reciente demuestra que el riesgo de las mujeres parece disminuir durante el embarazo y aumenta en los 9 meses tras el parto, especialmente en aquellas con genotipo de SQTL tipo 2, y por lo tanto propone mantener el tratamiento con bloqueadores beta (BB) durante ese periodo16.
En los estudios recientes obtenidos del Registro Internacional de SQTL, la presencia de síncope previo aparece como el predictor pronóstico más importante de posteriores eventos mayores (MS o parada cardiaca). Nuevamente, este efecto depende del tiempo, lo que reafirma la importancia de evaluar el riesgo de los pacientes de manera dinámica. En efecto, un síncope en los 2 años previos multiplica el riesgo en más de 2 veces respecto a la historia de un síncope más allá de los 2 años11,12. Igualmente, el número de episodios sincopales también parece tener un efecto acumulativo en el riesgo, especialmente durante la adolescencia y la edad adulta (tabla 1)11,12.
En los últimos años se ha comprobado que el estudio genético aporta, además de utilidad diagnóstica, cierta significación pronóstica en algunos casos de SQTL. Estudios iniciales observaron una mayor tasa de síncope, MS o parada cardiaca en los pacientes con genotipo de SQTL tipo 1 que en los de tipo 2 o tipo 317. Sin embargo, la mayor tasa de eventos se debía fundamentalmente a una mayor incidencia de síncopes. La incidencia de eventos con evolución fatal fue definitivamente superior en los pacientes con SQTL tipo 33,17. Actualmente, estudios con inclusión de mayor número de pacientes parecen confirmar que el tipo de SQTL no es tan determinante del pronóstico como lo son otros factores clínicos (fig. 3C)11–13. Únicamente ha quedado confirmado que el genotipo tipo 3 confiere un peor pronóstico más allá de los 40 años de edad, en los que multiplica casi hasta en 5 veces el riesgo de MS o parada cardiaca respecto a los otros subtipos de SQTL (tabla 1)14. Aunque poco frecuentes, es importante recordar que las formas Jervell y Lange-Nielsen se caracterizan por afección precoz y una tasa de eventos elevada, de forma que el 90% de los pacientes tienen síntomas en los primeros años de vida4.
Otros datos genéticos tienen relevancia clínica demostrada. En los pacientes con SQTL tipo 1, la presencia de una mutación localizada en un segmento transmembrana del gen KCNQ1 o de una mutación con efecto dominante negativo (esto es, que produce una disminución > 50% de la corriente IKs) se relaciona con un peor pronóstico y es factor independiente de MS o parada cardiaca (HR=2,26; IC del 95%, 1,56–3,25; p < 0,001) tras ajustar las variables clínicas (fig. 3D)18. En este mismo contexto se ha descrito una mutación relacionada con un fenotipo de extraordinaria malignidad, KCNQ1/A341V, caracterizada por la presencia de síntomas (síncope o MS) en hasta el 75% de casos, en edad joven, e intervalos QTc más largos19. Por su parte, entre los pacientes con SQTL tipo 2, la presencia de mutaciones que codifican para un segmento del poro del canal de potasio se han relacionado también con un peor pronóstico (el 74% de eventos cardiacos a lo largo de la vida frente al 35% en pacientes con mutaciones en otras regiones)20.
Vale la pena destacar que no se ha demostrado que la presencia de antecedentes familiares de MS indique mayor riesgo en ninguno de los estudios publicados hasta la fecha3,11–14.
TratamientoLa primera medida que tomar con todos los pacientes con SQTL es la abolición de situaciones desencadenantes de arritmias21 y la prohibición de toma concomitante de fármacos que alargan el intervalo QT; una lista exhaustiva de estos se puede encontrar en www.qtdrugs.org. Los BB son el tratamiento farmacológico de primera línea en los pacientes con SQTL, y actualmente se propone iniciarlos en todos los pacientes desde el momento en que son diagnosticados, especialmente si son SQTL tipo 1. Un estudio reciente demuestra que, en los pacientes con SQTL tipo 1 tratados con BB, la aparición de síntomas ocurre, en más del 90% de casos, en relación con la discontinuidad del tratamiento o toma concomitante de fármacos que alargan el intervalo QT22. Sin embargo, hasta un 23 y un 32% de los pacientes con SQTL tipo 2 y tipo 3, respectivamente, presentan síntomas a pesar de estar en tratamiento con BB23. El DAI es hoy la terapia indicada para pacientes considerados en alto riesgo, esto es, pacientes recuperados de una MS, pacientes que persisten sintomáticos pese a los BB, pacientes que presentan síntomas y/o intervalo QTc > 500 ms y tienen contraindicación para los BB y pacientes con formas Jervell y Lange-Niel-sen2. Algunos autores proponen también la implantación de DAI como primera terapia en pacientes con síntomas y QTc > 500 ms (junto al inicio de BB) y en los pacientes con SQTL tipo 3, especialmente si son varones3. La simpatectomía cervical también se ha utilizado como tratamiento antiadrenérgico en el SQTL tipo 1, pero su eficacia es escasa24. Recientemente se ha propuesto el tratamiento con fármacos bloquea-dores del sodio para los pacientes con SQTL tipo 3. Se ha demostrado que la flecainida mejora el intervalo QTc en estos pacientes, pero por el momento se desconoce si también mejora la incidencia de eventos25.
Síndrome de qt cortoEl SQTC es una entidad relativamente nueva, descrita por primera vez en el año 2000 en una familia con fibrilación auricular y un caso aislado complicado con MS26. Otras dos familias con alta incidencia de MS fueron descritas en 200327, y desde entonces se han comunicado en la literatura unos 50 casos. A pesar de los pocos casos conocidos, se han definido ya cinco formas distintas de SQTC, cada una de ellas producida por mutación en un gen distinto. La primera mutación relacionada con el síndrome fue identificada en el gen KCNH2, que codifica para el canal de corrientes IKr y da lugar al SQTC tipo 128. El SQTC tipo 2 se produce por mutaciones en el gen KCNQ1 de corrientes IKs29 y el SQTC tipo 3 responde a mutaciones en el gen KCNJ2 que codifica para las corrientes IK1. El efecto final en todos ellos es el aumento de las corrientes de salida de potasio, lo que acorta la repolarización y el intervalo QT en el ECG (fig. 1B I). Los SQTC tipos 4 y 5 se producen por mutaciones que causan hipofunción del canal de calcio (genes CAC-NB2b y CACNA1C respectivamente) y dan lugar a una forma combinada de SQTC y SBr (fig. 1B II)30. Los mecanismos que favorecen la aparición de arritmias en los pacientes con SQTC parecen ser la disminución del periodo refractario junto con el aumento de la dispersión transmural de la repolarización y disminución concomitante de la dispersión epicárdica31 (fig. 1B III).
Debido al bajo número de casos descritos hasta la fecha, los criterios diagnósticos de SQTC no están del todo definidos aún32. Estudios epidemiológicos señalan como anormalmente bajos los intervalos QTc < 360 ms en varones y < 370 ms en mujeres33. Sin embargo, en la serie más larga de SQTC descrita hasta la fecha, con 29 pacientes, todos ellos mostraron intervalo QT < 320 ms y QTc < 340 ms34, de forma que hoy se tiende a aceptar estos límites para el diagnóstico de la enfermedad. Característicamente, el ECG de los pacientes con SQTC muestra ondas T picudas y simétricas, aunque ello no se cumple en todos los casos ni en todos los genotipos4 (fig. 2B).
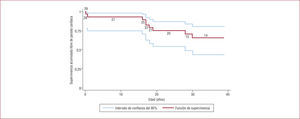
En la serie de 29 pacientes, más del 60% presentó síntomas y el 31%, en forma de MS, a menudo la primera manifestación de la enfermedad (fig. 4)34. Los síntomas aparecieron en algunos casos desde el primer año de vida, y en prácticamente el 25% de los casos se documentó fibrilación auricular asociada. Se piensa que la malignidad descrita puede deberse al sesgo que habitualmente existe en los primeros años que siguen a la descripción de una nueva enfermedad, cuando sólo se diagnostican los casos más agresivos.

Historia natural de los pacientes con síndrome de QT corto. Reproducido con autorización de Giustetto et al34.
Básicamente por la falta de datos, actualmente se sabe poco acerca de la estratificación del riesgo en el SQTC. La duración del intervalo QTc, el sexo, la inducibilidad de arritmias ventriculares en el estudio electrofisiológico (EEF) y el estudio genético no se han demostrado de valor predictivo en los pocos pacientes identificados32,34. Por lo tanto, y dada la alta incidencia de eventos descrita hasta la fecha, algunos autores han tomado por el momento la precaución de proteger con DAI a la mayoría de los pacientes diagnosticados32. Sin embargo, ello puede conllevar problemas, principalmente por sobresensado de las ondas T picudas32. Se ha demostrado que el tratamiento con quinidina alarga el intervalo QT, normaliza la morfología de la onda T, prolonga el periodo refractario ventricular y previene la inducibilidad de arritmias durante el EEF31,34. Por ello, se ha propuesto este tratamiento para pacientes no candidatos inicialmente a DAI (niños de corta edad), pacientes con múltiples episodios de FV como tratamiento coadyuvante al DAI, como tratamiento para la FA asociada y como posible terapia para las tormentas arrítmicas32.
Síndrome de brugadaFisiopatología y diagnósticoEl SBr fue descrito en 1992 como una entidad clínica caracterizada por un patrón de ECG típico en derivaciones precordiales derechas (elevación descendente del segmento ST de V1 a V3, con morfología similar al bloqueo de rama derecha) y una propensión aumentada a las arritmias ventriculares y la MS35. Hoy se calcula que el síndrome afecta a 1:5.000 individuos en el mundo occidental, y causa un 4–12% de todas las MS y hasta el 20% de las MS en corazón estructuralmente normal36.
El SBr fue relacionado inicialmente con mutaciones en el gen SCN5A37. En general, se halla una mutación en este gen en un 18–30% de los casos, y el efecto en todos ellos es la pérdida de función del canal de sodio, lo que produce un gradiente de corrientes positivas de salida (por predominio relativo de las corrientes Ito) durante la fase 1 del potencial de acción (fig. 1C I)36. Ello da lugar a dispersión de la repolarización transmural y epicárdica, base para la producción de arritmias ventriculares por un mecanismo de reentrada en fase 2 (fig. 1C III)36. En los últimos años se han identificado mutaciones en otros genes que también causan SBr y producen arritmias por un mecanismo similar38: GPD1-L, que produce hipofunción del canal de sodio de manera indirecta, CACNA1C y CACNB2b, que producen una pérdida de función del canal de calcio y dan lugar a un síndrome combinado con QT corto (fig. 1B II)30 y, muy recientemente, KCNE3, que produce secundariamente un aumento de las corrientes transitorias de potasio Ito (fig. 1C II)39.
Para establecer el diagnóstico de SBr, es necesario evidenciar el patrón ECG característico, conocido como tipo 1, sea de forma espontánea o tras provocación con un fármaco bloqueador del canal de sodio (fig. 2C)36, habitualmente en precordiales derechas, pero en ocasiones presente en derivaciones inferiores o incluso izquierdas40. En el último Consensus publicado en 2005, se definieron unos criterios diagnósticos según los cuales, además del patrón ECG, debería comprobarse la presencia de síntomas, taquicardia ventricular documentada o historia familiar del síndrome36. Hoy se tiende a considerar que la presencia sola de un ECG tipo 1 apunta a un paciente con fenotipo positivo y que, por lo tanto, requiere valoración y seguimiento independientemente de si cumple los otros criterios propuestos por el Consensus.
Historia natural y estratificación del riesgoLos pacientes con SBr permanecen en su mayoría asintomáticos. No obstante, un 17–42% sufre en algún momento de su vida síncope o MS como consecuencia de una arritmia cardiaca41–44. Esta cifra probablemente sobrestime la incidencia real de eventos, dado que una gran parte de los pacientes asintomáticos no son diagnosticados. La edad de presentación de los síntomas, especialmente de MS, se sitúa alrededor de la cuarta década de la vida38, sin que hasta la fecha se haya encontrado una explicación para ello.
Debido a la gran variabilidad fenotípica de los pacientes con SBr, la búsqueda de parámetros para ayudar a la estratificación del riesgo ha sido interés de primera línea en los últimos años41–44. Si bien la mayoría de los factores de riesgo han sido confirmados de manera independiente en las distintas series publicadas, hay algunas discrepancias45,46.
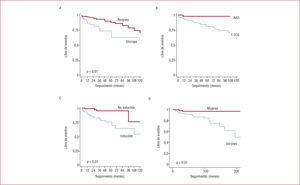
La presencia de síntomas previos es factor de riesgo reconocido por todos los estudios41–45. Datos de Brugada et al41 confirman que, entre los pacientes que han sufrido una MS recuperada, el 62% presenta una nueva arritmia en un periodo medio de 54 meses. Igualmente, Brugada et al45 demostraron, en su última serie procedente de la base de datos internacional constituida por 547 pacientes sin parada cardiaca previa, que la presencia de síncope aumenta en más de 2,5 veces la probabilidad de MS o fibrilación ventricular (FV) a los 2 años (fig. 5A). En la misma línea, Eckardt et al43 observaron, en una serie de 212 pacientes de menor riesgo seguidos durante una media de 40 meses, la aparición de eventos cardiacos en el 6% de los pacientes que previamente habían sufrido síncope, y sólo en un 1% de los pacientes previamente asintomáticos (p=0,028).
 en los pacientes con síndrome de Brugada. A: síntomas previos (reproducido con autorización de Brugada et al42). B: ECG tipo 1. C: inducibilidad en el EEF (reproducido con autorización de Brugada et al42). D: sexo masculino (reproducido con autorización de Benito et al47). A-C provienen del estudio con 547 pacientes sin antecedentes de parada cardiaca previa obtenidos de la base de datos internacional. D proviene de la serie de 384 pacientes de los dos centros de referencia participantes del estudio.")
Factores pronósticos (probabilidad de muerte súbita o fibrilación ventricular) en los pacientes con síndrome de Brugada. A: síntomas previos (reproducido con autorización de Brugada et al42). B: ECG tipo 1. C: inducibilidad en el EEF (reproducido con autorización de Brugada et al42). D: sexo masculino (reproducido con autorización de Benito et al47). A-C provienen del estudio con 547 pacientes sin antecedentes de parada cardiaca previa obtenidos de la base de datos internacional. D proviene de la serie de 384 pacientes de los dos centros de referencia participantes del estudio.
La presencia espontánea de un ECG tipo 1 también es factor pronóstico en los pacientes con SBr41–44. Dada la naturaleza dinámica del ECG en estos pacientes, para este criterio se suele considerar la documentación de un ECG tipo 1 espontáneo al menos una vez en la vida. En la misma serie de 547 pacientes de Brugada et al45, ello se relacionó con un riesgo incrementado frente a los pacientes que sólo mostraron ECG tipo 1 tras un test de provocación con un fármaco bloqueador del canal de sodio (HR=7,69; IC del 95%, 1,9–33,3) (fig. 5B). Sin embargo, esta variable no se ha confirmado como factor independiente de mal pronóstico en todos los estudios, principalmente porque el ECG tipo 1 espontáneo suele aparecer en pacientes sintomáticos y este parámetro tiene habitualmente superior significación pronóstica.
El papel del EEF en la estratificación del riesgo en el SBr es el tema que ha despertado más controversia en los últimos años45,46. En todas las series publicadas por el grupo de Brugada, la inducibilidad de FV durante la estimulación ventricular programada se ha demostrado de significación pronóstica, incluso por encima de otras variables, como el ECG41,42,45. En su serie de 547 pacientes, Brugada et al demostraron que la inducibilidad de arritmias en el EEF es factor independiente relacionado con un incremento de casi 6 veces en el riesgo de MS o FV durante el seguimiento (fig. 5C). Pero quizá una conclusión más llamativa de este estudio sea el hallazgo de que el EEF resultó la variable más potente para predecir eventos en los pacientes asintomáticos y sin historia familiar de SBr, los llamados casos fortuitos. En efecto, de los 167 pacientes fortuitos incluidos en este estudio, 11 presentaron eventos, y la inducibilidad de arritmias en el EEF fue el único factor independiente relacionado con ellos45. Otros autores no han podido confirmar estos resultados43,44,46, lo que se ha atribuido al uso de diferentes protocolos de estimulación ventricular, la inclusión de pacientes con dudoso diagnóstico y las bajas tasas de eventos comunicadas en otros estudios38.
Desde la descripción de las primeras series, se ha señalado la mayor agresividad del SBr en varones que en mujeres. Nuestro grupo ha analizado recientemente esta observación en 384 pacientes (272 varones y 112 mujeres), obtenidos únicamente de dos centros de referencia (Hospital Clínic de Barcelona y UZ Brussels) para evitar el gran sesgo de selección que se ha atribuido a la base de datos internacional, constituida principalmente por pacientes en alto riesgo47. En el momento de inclusión en el estudio, varones y mujeres mostraban características clínicas diferentes: mientras que los varones tenían más frecuentemente síntomas, ECG tipo 1 espontáneo y mayor inducibilidad en el EEF, las mujeres mostraban más propensión a alteraciones de la conducción durante la administración de fármacos bloqueadores del sodio. El pronóstico de ambas poblaciones fue también marcadamente diferente. De los 31 eventos (MS o FV) documentados en toda la población en un seguimiento medio de 58 meses, 28 ocurrieron en varones (lo que representa el 11,6%) y 3 en mujeres (el 2,8%) (p=0,003) (fig. 5D)47. Los factores de riesgo descritos para poblaciones mixtas (síntomas, ECG tipo 1 espontáneo e inducibilidad en el EEF) se confirmaron como válidos para estratificar el riesgo en los varones. Por el contrario, en las mujeres, los trastornos de la conducción parecieron relacionarse mejor con la tasa de eventos47. Se ha hipotetizado que las diferencias en el comportamiento entre varones y mujeres responden en parte a influencias hormonales y a las diferencias en la densidad epicárdica de corrientes Ito entre uno y otro sexo47.
Al igual que ocurría con el SQTL, los antecedentes familiares de MS no parecen marcar el pronóstico de los pacientes con SBr. La presencia de mutación identificada en el gen SCN5A por sí sola tampoco se relaciona con una mayor tasa de eventos. Recientemente, nuestro grupo ha analizado si el tipo de mutación y su localización en el gen SCN5A pueden aportar datos sobre el pronóstico de los pacientes con SBr y mutación identificada. Datos preliminares indican que las mutaciones que dan lugar a una proteína truncada podrían aumentar el riesgo en este subgrupo de pacientes48.
TratamientoEl único tratamiento de eficacia claramente demostrada en el SBr es la implantación de DAI. De acuerdo con lo explicado, deberían recibir DAI todos los pacientes sintomáticos (con MS recuperada o síncope previo) y los pacientes asintomáticos si muestran inducibilidad en el EEF, especialmente si presentan espontáneamente un ECG tipo 136. De todos los fármacos ensayados hasta la fecha, la quinidina, por su efecto bloqueador de las corrientes Ito, ha mostrado eficacia en ciertos contextos, como en el tratamiento de la tormenta arrítmica o como terapia adyuvante al DAI en pacientes con múltiples descargas, y se ha propuesto como alternativa al DAI para el tratamiento de niños con alto riesgo38.
Taquicardia ventricular catecolaminérgicaEl primer caso de taquicardia bidireccional en relación con el ejercicio se comunicó en 1975, y a continuación se describieron 4 casos más de taquicardia ventricular (TV) inducida por catecolaminas49. En 1995 se publicó la primera serie de 21 niños con TVPC, todos ellos con síncope relacionado con estrés o emoción intensa y sin evidencia de enfermedad cardiaca estructural ni intervalo QT alargado en el ECG50. Característicamente, en todos los casos se desencadenó de manera reproducible TV polimórfica tras el ejercicio, en 15 de ellos en forma de TV bidireccional (con cambio de eje de latido a latido). Posteriores series algo más extensas, y sobre todo la identificación de varios genes causales, han permitido ampliar nuestro conocimiento sobre esta nueva entidad. Actualmente se distinguen dos formas de TVPC51: tipo 1, de transmisión autosómica dominante y producida por mutaciones en el gen RyR2, el receptor de la rianodina que se encuentra en la membrana del retículo sarcoplásmico (RS), y tipo 2, de transmisión recesiva y producido por mutaciones en el gen que codifica para la isoforma cardiaca de calsecuestrina (CASQ2), principal reservorio de calcio en el RS (fig. 1D I). El efecto por uno u otro mecanismo es la liberación exagerada de calcio desde el RS al citoplasma en presencia de estimulación adrenérgica, lo que puede favorecer secundariamente la aparición de pospotenciales tardíos y desencadenar la arritmia (fig. 1D II)51. Mutaciones en uno de estos dos genes pueden hallarse en un 50–70% de los pacientes con TVPC, lo que indica que deben existir otros genes implicados aún no descritos.
Los pacientes con TVPC suelen presentar un ECG basal normal. Los síntomas y la evidencia de arritmias aparecen típicamente durante el ejercicio, de forma que la prueba de esfuerzo suele ser la herramienta diagnóstica de mayor utilidad. Una característica importante, que a menudo ayuda a establecer la distinción con el SQTL, es que las arritmias suelen aparecer alrededor de los 110–130 lpm, dato relativamente constante para todos los pacientes, e inicialmente en forma de extrasístoles ventriculares que van haciéndose más frecuentes hasta desencadenar taquicardia bidireccional, polimórfica o FV con progresivas cargas de ejercicio50,52.
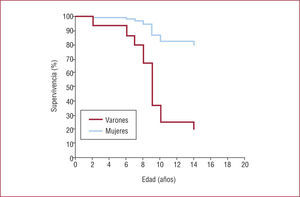
En la serie más amplia publicada, con 30 pacientes probandos y 9 familiares afectos, Priori et al52 comprobaron que la penetrancia en la TVPC, como en otras enfermedades eléctricas primarias, es incompleta (alrededor del 15% de los pacientes eran portadores silentes). Sin embargo, la mayoría de los pacientes sufrieron síntomas, habi-tualmente durante la infancia pero en algunos casos también durante la edad adulta. De hecho, el pronóstico sin tratamiento es ominoso, con una tasa altísima de MS en las primeras décadas de la vida50,52. En la serie de Priori et al52, el único factor de riesgo identificado fue el sexo masculino, especialmente entre los portadores de mutación en el gen RyR2 (varones frente a mujeres, HR=4,2; IC del 95%, 1,2–15) (fig. 6). La presencia de mutación no implicó ninguna significación pronóstica. Por su parte, el EEF, en esta y otras series50, no ha aportado utilidad en el diagnóstico ni en la estratificación del riesgo, ya que la mayoría de los pacientes no son inducibles.

Pronóstico de los pacientes con taquicardia ventricular polimórfica catecolaminérgica en función del sexo. Reproducido con autorización de Priori et al52.
Los pacientes con TVPC deben restringir al mínimo la actividad deportiva y evitar sobresaltos emocionales21. El tratamiento de primera línea, que debe iniciarse en todos los pacientes diagnosticados como prevención primaria, son los BB, que actúan inhibiendo la descarga adrenérgica desencadenante de las arritmias. Sin embargo, más del 45% de los pacientes presentan TV sostenida o FV estando en tratamiento con BB, y hasta el 19% se complica con MS52,53. Algunos autores han propuesto el tratamiento con verapamilo, que no ha obtenido mejores resultados53. Trabajos recientes con pocos pacientes han propuesto nuevas estrategias, como el uso simultáneo de ambos fármacos54 o la simpatectomía cervical55, aunque se requieren estudios más a largo plazo y con mayor número de pacientes para establecer su papel definitivo. El DAI se ha demostrado eficaz en los pacientes con TVPC, y está indicado como prevención secundaria para todos los pacientes y como prevención primaria en aquellos con síntomas o evidencia de taquicardia pese a tratamiento con BB a las dosis máximas toleradas51.
ConclusionesEl SQTL, el SQTC, el SBr y la TVPC se encuadran entre las enfermedades arritmogénicas hereditarias sin cardiopatía estructural. Debido a su penetrancia incompleta y su variabilidad fenotípica, que alberga desde la ausencia de síntomas hasta la MS en edad temprana, es fundamental una correcta estratificación del riesgo de los pacientes que sufren alguna de estas enfermedades. En el SQTL, los principales factores de riesgo son un intervalo QTc > 500 ms, la presencia de síncope previo, el sexo (dependiendo de la edad), las formas Jervell y Lange-Nielsen y algunos datos genéticos como las mutaciones con dominancia negativa (en el SQTL tipo 1) o las mutaciones del poro del canal (en el SQTL tipo 2). En el SBr, son variables relacionadas con un mal pronóstico la presencia de síntomas previos, un ECG tipo 1 de forma espontánea, la inducibilidad de arritmias ventriculares en el EEF y el sexo masculino. Básicamente por el bajo número de pacientes descritos, se conocen pocos datos acerca de la estratificación del riesgo en el SQTC y la TVPC, aunque en esta el sexo masculino parece conferir un peor pronóstico.
FinanciaciónFinanciado por el Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica (I+D+I), Instituto de Salud Carlos III/ FIS (ref. CM06/00189) y la Sociedad Española de Cardiología: Beca para formación en centros extranjeros, ambas en España.
Conflicto de interesesNinguno.