La muerte súbita (MS) de personas jóvenes suele tener una causa genética, por lo cual la «autopsia molecular» puede tener implicaciones importantes para los familiares. El objetivo del estudio es evaluar el rendimiento diagnóstico de un programa de autopsia molecular mediante secuenciación masiva.
MétodosEstudio prospectivo de una cohorte de pacientes consecutivos de edad ≤ 50 años y fallecidos por MS no violenta, a los que se realizó autopsia molecular mediante paneles amplios por secuenciación masiva, con posterior cribado familiar clínico y genético. Se analizan datos demográficos, clínicos, toxicológicos y genéticos.
ResultadosSe estudiaron 123 casos consecutivos de MS a edades ≤ 50 años. La incidencia de MS fue de 5,8 casos/100.000 individuos/año, a una media de edad de 36,15±12,7 años; 95 (77%) eran varones. La causa fue cardiaca en el 53%; MS inexplicada en el 24%, tóxicos en el 10,6% y MS del lactante en el 4%. De las cardiacas, el 38% por cardiopatía isquémica, el 7% por miocardiopatía arritmogénica, el 5% por miocardiopatía hipertrófica y el 11% por hipertrofia ventricular izquierda idiopática. Se indicó análisis genético en 62 casos (50,4%). Se hallaron variantes genéticas en 42 (67,7%), con una media de 3,4±4 variantes/paciente, que se consideraron patogénicas o probablemente patogénicas en el 30,6%. De las MS inexplicadas, hasta el 70% presentó alguna variante genética. El estudio familiar permitió detectar a 21 portadores o afectos, 5 de ellos estaban en riesgo, por lo que se indicó implante de desfibrilador.
ConclusionesEl estudio protocolizado y exhaustivo de la MS cardiaca de personas jóvenes es factible y necesario. En un alto porcentaje la causa es genética y, por lo tanto, existen familiares en riesgo que pueden beneficiarse de un diagnóstico y un tratamiento precoces para evitar complicaciones.
Palabras clave
La muerte súbita (MS) se define como el fallecimiento inesperado de una persona, aparentemente sana o con alguna enfermedad conocida, en la primera hora tras el comienzo de los síntomas o cuando se la haya visto por última vez aparentemente sana en las últimas 24 h1. En la mayoría de los casos es de origen cardiaco2. Es la principal causa de muerte en el mundo industrializado, con una prevalencia del 20%3, y tiene un efecto psicosocial devastador en las familias de las víctimas y en los medios, sobre todo si ocurre a personas jóvenes. En la mayoría de los casos, cuando estos fallecimientos afectan a individuos de edad <35-40 años, se deben a enfermedades cardiacas con una causa genética subyacente (miocardiopatías o canalopatías), lo que implica que puede haber más familiares en riesgo, a diferencia de los individuos de más edad, que fallecen principalmente de cardiopatía isquémica2,4.
El estudio de los casos de MS recae en su mayoría dentro del ámbito de la medicina forense5,6.
Al estudio histológico completo del corazón más el estudio genético guiado por la histopatología, se lo ha denominado «autopsia molecular», y con ella se puede llegar a saber la causa final de la MS en un alto porcentaje de casos7,8. Así, ante el fallecimiento de una persona joven, además de la preceptiva autopsia judicial, es conveniente realizar una autopsia molecular en casos de miocardiopatías, autopsia clínica normal o hallazgos inespecíficos8, algo que desgraciadamente está muy poco extendido tanto en España como en otros países5,9.
El término síndrome de MS inexplicada (sudden unexplained death syndrome [SUDS]) se utiliza para los casos de MS de etiología no filiada pese a una autopsia y un estudio toxicológico adecuados10, y representa aproximadamente el 40% de todas las MS de pacientes de 1 a 35-40 años6,11,12. Se estima que en torno al 10-25% de las MS inexplicadas del adulto y hasta un tercio de las MS infantiles y juveniles pueden ser causadas por canalopatías (síndrome de QT largo, síndrome de QT corto, síndrome de Brugada o taquicardia ventricular polimórfica catecolaminérgica)2,13,14, y en estos casos la rentabilidad diagnóstica (entendida como el beneficio adicional obtenido) del estudio genético por secuenciación masiva (NGS) es del 29-32%15,16.
La incidencia anual de MS en las diversas series publicadas varía bastante, entre otras cosas por las diferencias en la franja de edad considerada. Se estima que entre 1 y 35-40 años varía de 1,3 a 8,5/100.000 personas-año6,11,17–19.
El objetivo del presente estudio es analizar la rentabilidad de la aplicación secuencial de una batería de pruebas diagnósticas (clínicas, histológicas y toxicológicas), que incluye el análisis genético mediante los paneles de NGS más amplios existentes y un posterior estudio familiar en cascada (estudio clínico y genético), en el diagnóstico de la MS de personas jóvenes.
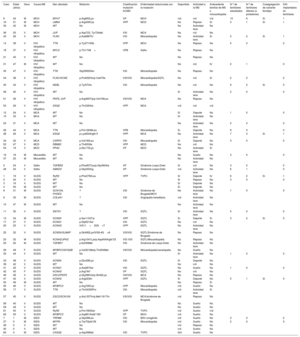
MÉTODOSSe trata de un programa asistencial iniciado en febrero de 2015 y que se mantiene hasta la fecha. Se diseñó un programa colaborativo multidisciplinario en las Islas Baleares entre los hospitales y el Instituto de Medicina Legal para el estudio completo de los casos de MS de personas jóvenes. Este instituto concentra todos los casos de MS ocurridos en las Islas Baleares, que abarca una población de 1.144.392 habitantes (Instituto Nacional de Estadística, 2016). Se formó un grupo de trabajo entre cardiólogos, forenses, patólogos, intensivistas, genetistas, biólogos y químicos, con el objetivo de protocolizar el estudio de la MS no traumática de personas de edad ≤ 50 años, que consistía en la realización de una autopsia completa en todos los casos, con estudio exhaustivo anatomopatológico y genético, intentando llegar al diagnóstico causal de estas MS. El programa se denomina MUSIB (MUerte Súbita Islas Baleares).
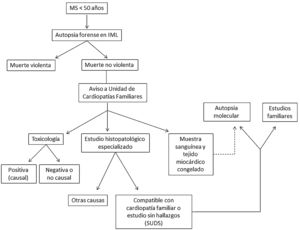
La autopsia judicial se realizó en el Instituto de Medicina Legal de las Islas Baleares según el protocolo estándar basado en las guías forenses8. El corazón y otras vísceras se enviaron al Instituto Nacional de Toxicología y Ciencias Forenses de Barcelona, centro de referencia nacional, para su estudio macroscópico, microscópico y toxicológico. Además, se remitieron muestras de sangre al laboratorio de genética del Hospital Universitario Son Espases para el posterior análisis molecular, guiado por los resultados de la histopatología. El estudio se coordinó desde la Unidad de Cardiopatías Familiares del Hospital Universitario Son Llàtzer (figura 1).

Se recogieron datos clínicos y familiares, circunstancias de la muerte, electrocardiogramas previos (si estaban disponibles), tratamientos previos, resultados de histopatología macroscópica y microscópica y toxicología. Posteriormente se realizó estudio genético mediante NGS de todos los casos con diagnóstico de miocardiopatía en la histopatología, hallazgos inespecíficos o ausencia de cardiopatía estructural macroscópica y microscópica (SUDS). En los casos de miocardiopatías, se estudiaron paneles amplios de genes relacionados con la miocardiopatía hallada. En los casos de SUDS o hallazgos histopatológicos inespecíficos se secuenciaron por NGS paneles de entre 194 y 380 genes. Hasta 2017, se externalizaba el análisis de las muestras a los laboratorios Health in code (A Coruña), Imegen (Valencia) y Centogene (Alemania), y después se han analizado en el laboratorio de genética del Hospital Universitario Son Espases. Las variantes se filtraron utilizando un protocolo de priorización preestablecido, principalmente basado en el presumible impacto funcional de la proteína y en la frecuencia alélica. Se utilizaron asimismo herramientas informáticas de predicción in silico. Se reanalizó la patogenicidad de todas las variantes incluidas de acuerdo con las recomendaciones vigentes del American College of Medical Genetics and Genomics y la Association for Molecular Pathology (ACMG/AMP) y se excluyeron las consideradas no patogénicas o posiblemente no patogénicas y aquellas de significado incierto que presentaran frecuencias ≥ 0,02% en las bases de datos de GnomAD, ClinVar y Exac o sin cosegregación demostrada en los casos estudiados20. Los hallazgos de variantes genéticas raras se confirmaron mediante secuenciación Sanger. Asimismo, como el protocolo conlleva el estudio familiar clínico y genético en cascada (se inicia el estudio de todos los familiares de 1.er grado y se prosigue con los de 2.o, 3.er grado, etc., si se hallan portadores), se estudió la cosegregación de las variantes halladas en todos los casos en que ha sido posible.
Los datos se trataron con confidencialidad siguiendo la normativa vigente. El programa fue aprobado por el CEIC de las Islas Baleares.
Análisis estadísticoSe realizó un estudio descriptivo mediante el cálculo de frecuencias de las variables cualitativas, así como la media±desviación estándar o la mediana [intervalo intercuartílico] para las variables cuantitativas. Se utilizó el paquete estadístico SPSS 15.0.
RESULTADOSSe incluyó en el estudio un total de 123 casos consecutivos de MS que ya tenían todo el estudio completo histopatológico, químico y genético, si bien hasta la fecha los casos de MS incluidos en el programa MUSIB fueron 183 en total (los casos no analizados ahora no tienen todas las pruebas concluidas). La incidencia en las Islas Baleares de MS de personas de edad ≤ 50 años está en 5,8 casos/100.000 individuos/año. Por sexo, la incidencia es mucho mayor en los varones (10,1 frente a 1,6). La media de edad fue 36,15±12,7 (intervalo, 0-50) años, 95 eran varones (77%) y 28, mujeres (23%). La media de edad de los varones fue 37,5±11,4 años y la de las mujeres, 31,5±15,6.
La prevalencia de la MS varió en relación con la edad: la mayoría de los casos se concentran entre los 41 y los 50 años (58 casos; 47%). Los casos entre los 31 y los 40 años fueron 32 (26%); entre los 21 y los 30, 20 (16,2%); entre los 11 y los 20, 6 (4,9%), y a edad ≤ 10 años, 7 casos (5,7%), de los que 5 eran lactantes. En cuanto al sexo, hubo más varones en todas las franjas de edad, salvo en lactantes.
En cuanto a las circunstancias de la MS, lo más frecuente fue en reposo (28,5%), seguido de MS durante una actividad diaria leve o cotidiana (25,2%), el sueño (15,4%) y el deporte (12,2%). En 23 casos (18,7%) la circunstancia de la MS fue desconocida. Solo en 13 (10,6%) había antecedentes familiares conocidos de MS o miocardiopatía. Se realizó reanimación cardiopulmonar avanzada en 65 casos (52,8%). Practicaban deporte habitualmente (amateurs o profesionales) 22 (17,9%): en 13 casos (59%) el fallecimiento ocurrió durante el ejercicio; en 5 (22,7%), durante una actividad diaria leve, y en 3 (13,6%), en reposo o durante el sueño.
Resultados toxicológicosSe detectó el consumo de alguna droga o fármaco en el 51,4%, si bien solo en 13 casos (10,6%) fue claramente la causa de la MS. Un 69% había consumido algo de alcohol; el 13%, cocaína; el 8%, cannabis; el 5%, 6-monoacetilmorfina y el 2%, 3,4-metilendioximetanfetamina. Además se hallaron con frecuencia fármacos (principalmente benzodiacepinas), pero en la mayoría a concentraciones no tóxicas.
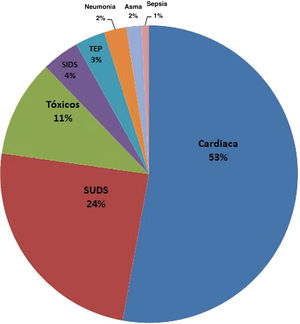
Resultados histopatológicosLa histopatología definió la MS como cardiaca en 95 casos: 65 (52,8%) con cardiopatía estructural y 30 (24,4%) con autopsia blanca (SUDS), y en 13 (10,6%) se debió a tóxicos. Otras causas menos frecuentes fueron: MS del lactante (sudden infant death syndrome [SIDS]) (4,1%), tromboembolia pulmonar (3,2%), neumonía (2,4%), asma (1,6%) y sepsis (0,8%) (figura 2).

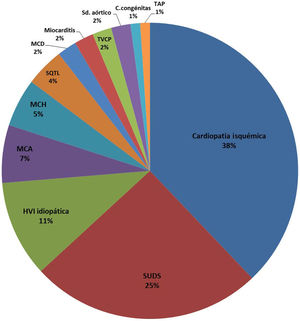
Más específicamente, desglosando las causas cardiacas (n=95, histopatología positiva+SUDS), en 36 (38%) se debió a cardiopatía isquémica; en 6 (7%), a miocardiopatía arritmogénica; en 5 (5%), a miocardiopatía hipertrófica; en 10 (11%), a hipertrofia ventricular izquierda idiopática; en 30 (31%), a SUDS (finalmente un 25%, porque la autopsia molecular permitió diagnosticar síndrome de QT largo en 4 casos y taquicardia ventricular catecolaminérgica polimórfica en 2), y otros casos menos frecuentes: miocardiopatías dilatadas, 2 (2%); miocarditis, 2 (2%); síndromes aórticos agudos, 2 (2%); cardiopatía congénita compleja con bloqueo auriculoventricular, 1 (1%), y taponamiento cardiaco agudo, 1 (1%) (figura 3).
. HVI: hipertrofia ventricular izquierda; MCA: miocardiopatía arritmogénica; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; SUDS: síndrome de muerte súbita inexplicada; SQTL: síndrome de QT largo; TAP: taponamiento agudo pericárdico; TVPC: taquicardia ventricular polimorfa catecolaminérgica.")
Causas cardiacas de MS (n=95, histopatología positiva+casos de SUDS). HVI: hipertrofia ventricular izquierda; MCA: miocardiopatía arritmogénica; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; SUDS: síndrome de muerte súbita inexplicada; SQTL: síndrome de QT largo; TAP: taponamiento agudo pericárdico; TVPC: taquicardia ventricular polimorfa catecolaminérgica.
Los casos de SUDS fueron un total de 30 (el 66,7% varones), a una media de edad de 36,7±10,9 años. El 30% eran deportistas. Las circunstancias de la MS fueron: el 23,3% durante el sueño, el 20% en reposo, el 23,3% en una actividad diaria leve y el 23,3% haciendo deporte. Tenían antecedente familiar de MS o miocardiopatía el 13,3%.
Resultados genéticosSe realizó análisis genético en 62 casos (50,4%) (tabla 1): a todos los casos de causa cardiaca (excepto el de cardiopatía congénita y el de taponamiento) y los 5 SIDS. Se hallaron una media de 3,4±4 variantes genéticas/paciente en un total de 42 pacientes (67,7%). Se catalogó la variante de significado incierto (VSI), VP o VPP en 40 casos (rendimiento del 64,5%). Si se consideran solo las VP o VPP, el rendimiento fue del 30,6%.
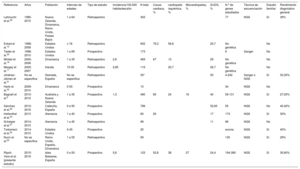
Pacientes con MS y estudio genético realizado: datos clínicos, genéticos y familiares, ordenados por la causa de la MS
| Caso | Edad (años) | Sexo | Causa MS | Gen afectado | Mutación | Clasificación mutación ACMG | Enfermedad relacionada con la mutación | Deportista | Actividad a la MS | Antecedente familiar de MS o miocardiopatía | N.o de familiares estudiados | N.o de familiares afectos (o portadores) | Cosegregación de variante-fenotipo | DAI implantados en familiares |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 42 | M | MCH | MYH7 | p.Arg652Lys | VP | MCH | n/d | n/d | n/d | 10 | 6 | Sí | 1 |
| 16 | 31 | M | MCH | LMNA | p.Arg439Cys | VPP | MCD | No | Reposo | Sí | 3 | 1 | Sí | 1 |
| 30 | 42 | M | MCH | WT | No | No | Actividad leve | No | ||||||
| 36 | 25 | V | MCH | JUP | p.Asp723_Tyr724del | VSI | MCA | No | n/d | No | ||||
| 43 | 26 | V | MCH | FLNC | p.Ala688Thr | VSI | Miocardiopatía | No | Actividad leve | Sí | 5 | 2 | Sí | 1 |
| 14 | 39 | V | HVI idiopática | TTN | p.Tyr27100fs | VPP | MCH | No | Reposo | No | 5 | 0 | 0 | |
| 18 | 37 | V | HVI idiopática | MYLK | p.Thr1748= | VPB | SdAo | No | Reposo | No | ||||
| 21 | 45 | V | HVI idiopática | WT | No | No | Reposo | No | ||||||
| 31 | 47 | M | HVI idiopática | WT | No | No | n/d | Sí | 2 | 1 | 0 | |||
| 35 | 47 | V | HVI idiopática | TTN | Gly25650Ser | VSI | Miocardiopatía | No | Reposo | No | ||||
| 44 | 38 | V | HVI idiopática | FLNC/KCNE | p.Pro632His/p.Val47Ile | VSI/VSI | Miocardiopatía/SQTL | No | n/d | Sí | 2 | 0 | 0 | |
| 45 | 43 | V | HVI idiopática | NEBL | p.Tyr57His | VSI | Miocardiopatía | No | n/d | No | 9 | 2 | Sí | 0 |
| 48 | 40 | V | HVI idiopática | WT | No | Sí | Actividad leve | No | 2 | 0 | 0 | |||
| 51 | 36 | V | HVI idiopática | PKP2, JUP | p.Arg490Trp/p.Val159Leu | VSI/VSI | MCA | No | Reposo | No | ||||
| 53 | 29 | V | HVI idiopática | DSG2 | p.Thr335Ala | VPP | MCA | n/d | n/d | n/d | ||||
| 12 | 24 | V | MCA | WT | No | Sí | Deporte | No | 1 | 0 | 0 | |||
| 15 | 33 | V | MCA | WT | No | Sí | Actividad leve | No | ||||||
| 24 | 31 | V | MCA | WT | No | No | Actividad leve | No | 2 | 0 | 0 | |||
| 26 | 44 | V | MCA | TTN | p.Pro12648Leu | VPB | Miocardiopatía | Sí | Deporte | No | 9 | 0 | 0 | |
| 28 | 26 | V | MCA | DSG2 | p.Lys834Argfs*3 | VPP | MCA | No | Actividad leve | No | 7 | 3 | Sí | 1 |
| 34 | 32 | V | MCA | CSRP3 | p.Val190Leu | VSI | Miocardiopatía | Sí | Deporte | No | 3 | 0 | 0 | |
| 52 | 47 | V | MCD | RBM20 | p.Thr653Ile | VPP | MCD | No | n/d | No | ||||
| 54 | 19 | V | MCD | PPA2 | p.Glu172Lys | VP | MCD | No | Actividad leve | Sí | ||||
| 19 | 18 | M | Miocarditis | WT | No | No | Sueño | No | 3 | 0 | 0 | |||
| 37 | 25 | M | Miocarditis | WT | No | No | Actividad leve | No | ||||||
| 5 | 24 | V | SdAo | TGFBR2 | p.Phe467Cys/p.Gly490Ala | VP | Síndrome Loeys-Dietz | Sí | n/d | No | 3 | 0 | 0 | |
| 49 | 43 | V | SdAo | SMAD3 | p.Gly245Arg | VP | Síndrome Loeys-Dietz | n/d | Actividad leve | No | 6 | 1 | 0 | |
| 1 | 19 | V | SUDS | RyR2 | p.Phe3790Leu | VPP | TVPC | Sí | Deporte | Sí | 6 | 2 | Sí | 1 |
| 2 | 24 | V | SUDS | WT | No | Sí | Reposo | No | 3 | 0 | 0 | |||
| 3 | 16 | V | SUDS | WT | No | Sí | Deporte | No | ||||||
| 4 | 50 | M | SUDS | WT | No | Sí | Deporte | No | ||||||
| 8 | 31 | M | SUDS | SCN10A, FHOD3 | ? | VSI | Síndrome de Brugada/MCH | No | Actividad leve | No | 1 | 0 | 0 | |
| 9 | 33 | M | SUDS | COL4A1 | ? | VSI | Angiopatía hereditaria | n/d | Actividad leve | No | ||||
| 10 | 47 | M | SUDS | WT | No | No | Actividad leve | No | ||||||
| 11 | 32 | V | SUDS | SNTA1 | ? | VSI | SQTL | Sí | Actividad leve | No | 5 | 0 | 0 | |
| 13 | 34 | M | SUDS | SCN5A | p.Ser1103Tyr | VPP | SQTL | Sí | Deporte | Sí | 3 | 2 | Sí | 0 |
| 17 | 37 | V | SUDS | KCNQ1 | p.Gly621Ser | VSI | SQTL | No | n/d | No | ||||
| 20 | 25 | V | SUDS | KCNH2 | IVS11+20G>T | VPP | SQTL | No | Actividad leve | No | ||||
| 22 | 32 | V | SUDS | SCN5A/SLMAP | p.Gln692Lys/IVS8-4G>A | VSI/VSI | SQTL/Síndrome de Brugada | No | Reposo | No | ||||
| 23 | 32 | M | SUDS | KCNH2/JPH2 | p.Arg1041Lys/p.Asp454Argfs*23 | VSI /VSI | SQTL/Miocardiopatía | No | Reposo | No | ||||
| 25 | 22 | M | SUDS | TGFBR1 | p.Ile339Met | VSI | Síndrome de Loeys-Dietz | No | Actividad leve | No | ||||
| 29 | 48 | F | SUDS | MYBPC3/SCN2B | p.Val321Met/p.Thr204Met | VSI/VSI | Miocardiopatía/canalopatía | No | Sueño | No | ||||
| 32 | 44 | V | SUDS | WT | No | Sí | Actividad leve | No | 1 | 0 | 0 | |||
| 33 | 48 | V | SUDS | SCN5A | p.Glu428Lys | VSI | SQTL | Sí | Deporte | No | ||||
| 39 | 42 | V | SUDS | WT | No | No | n/d | No | ||||||
| 41 | 22 | V | SUDS | KCNQ1 | p.Ala287Ser | VSI | SQTL | No | Sueño | No | 1 | 0 | 0 | |
| 42 | 40 | F | SUDS | KCNH2 | p.Arg744* | VP | SQTL | No | n/d | No | ||||
| 46 | 36 | V | SUDS | DSC2/PKP2 | p.Gly286Val/p.Gln62Lys | VSI/VSI | MCA | No | Reposo | No | ||||
| 47 | 42 | V | SUDS | KCNH2 | p.Arg22Gln | VPP | SQTL | Sí | Deporte | No | 5 | 1 | Sí | 0 |
| 50 | 49 | M | SUDS | WT | No | No | Reposo | Sí | ||||||
| 55 | 46 | V | SUDS | MYBPC3 | p.Arg726Cys | VPP | Miocardiopatía | n/d | Sueño | No | ||||
| 56 | 17 | V | SUDS | TTN | p.Thr34393Pro | VSI | Miocardiopatía | n/d | Actividad leve | Sí | ||||
| 57 | 45 | V | SUDS | DSC2/SCN10A | p.Ala133Thr/p.Met1161Thr | VSI/VSI | MCA/síndrome de Brugada | n/d | Reposo | No | ||||
| 58 | 40 | V | SUDS | WT | No | No | Sueño | No | ||||||
| 59 | 49 | V | SUDS | WT | No | No | Sueño | No | ||||||
| 61 | 50 | V | SUDS | RyR2 | p.Pro1583Ser | VPP | TVPC | n/d | Sueño | n/d | ||||
| 62 | 50 | V | SUDS | MYBPC3 | p.Arg891Alafs*160 | VP | MCH | n/d | Sueño | No | ||||
| 7 | 1 | M | SIDS | TRPM4 | p.Gly298Leu | VSI | BAV congénito | n/d | Sueño | No | 3 | 0 | 0 | |
| 27 | 0 | M | SIDS | MYPN | p.Trp7Glyfs*26 | VSI | Miocardiopatía | n/d | Sueño | No | 2 | 0 | 0 | |
| 38 | 0 | V | SIDS | WT | No | n/d | Reposo | No | ||||||
| 40 | 0 | V | SIDS | WT | No | n/d | Sueño | No | ||||||
| 60 | 0 | M | SIDS | CASQ2 | p.Asp398del | VSI | TVPC | N/d | Sueño | No |
ACMG: American College of Medical Genetics and Genomics; BAV: bloqueo auriculoventricular; DAI: desfibrilador automático implantable; HVI: hipertrofia ventricular izquierda; M: mujer; MCA: miocardiopatía arritmogénica; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MS: muerte súbita; n/d: información no disponible; NGS: estudio genético por secuenciación masiva; SUDS: síndrome de muerte súbita inexplicada; SdAo: síndrome aórtico; SIDS: síndrome de muerte súbita del lactante; SQTL: síndrome de QT largo; TVPC: taquicardia ventricular polimorfa catecolaminérgica; V: varón; VP: variante patogénica; VPB: variante probablemente benigna; VPP: variante probablemente patogénica; VSI: variante genética de significado incierto; WT: wild type (ausencia de mutaciones).
En las miocardiopatías, la rentabilidad del análisis genético fue la siguiente: de 6 casos de miocardiopatía arritmogénica, se hallaron variantes en 2; de 5 casos de miocardiopatía hipertrófica, en 4; de 10 casos de hipertrofia ventricular idiopática, en 6, y de 2 casos de miocardiopatía dilatada, 1 variante cada uno (tabla 1).
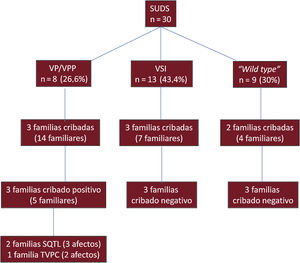
En los SUDS, se realizó análisis genético en todos los casos, y este permitió encontrar VSI, VP o VPP en 21 (70%): 15 variantes relacionadas con canalopatías (10 de síndrome de QT largo, 3 de síndrome Brugada y 2 de taquicardia ventricular polimorfa catecolaminérgica) y 10 con genes estructurales (tabla 1). Si se tiene en cuenta solo las VP o VPP, el rendimiento es del 26,6%.
Respecto a los casos de SIDS, se han estudiado 5 casos, 4 de ellos ocurridos durante el sueño. Se halló alguna variante en 3 casos, todas VSI, 2 en relación con canales iónicos (1 taquicardia ventricular catecolaminérgica, 1 bloqueo auriculoventricular) y 1 en relación con miocardiopatía (tabla 1).
Resultados del estudio familiarSe indicó estudio familiar a todos los que tuvieran estudio genético realizado, y acudieron a consulta 26 familias y 104 individuos (media, 4/familia); hasta el momento se ha diagnosticado a 21 portadores o afectos. De estos, se ha indicado implante de desfibrilador como prevención primaria, con base en la estratificación de riesgo, a 5 familiares: 3 miocardiopatías hipertróficas, 1 miocardiopatía arritmogénica y 1 taquicardia ventricular catecolaminérgica. Uno de ellos ha tenido ya una descarga apropiada.
En los SUDS se ha estudiado hasta la fecha a 25 familiares;por el momento se ha encontrado a 3 afectos de síndrome de QT largo y 2 de taquicardia ventricular polimorfa catecolaminérgica (figura 4).

Diagrama de flujo de la serie de pacientes con MS y diagnóstico necrópsico de SUDS: rendimiento del análisis genético y del cribado familiar. MS: muerte súbita; NGS: estudio genético por secuenciación masiva; SIDS: síndrome de muerte súbita del lactante; SUDS: síndrome de muerte súbita inexplicada; VP: variante patogénica; VPP: variante probablemente patogénica; VSI: variante de significado incierto; SQTL: síndrome de QT largo; TVPC: taquicardia ventricular polimorfa catecolaminérgica.
El estudio familiar ha permitido demostrar la cosegregación de varias variantes genéticas y, por lo tanto, confirmar su patogenicidad. Es el caso de las variantes p.Ser1103Tyr en el gen SCN5A (VPP), que produce QT largo; p.Phe3790Leu en el gen RyR2 (VPP), que produce taquicardia ventricular polimorfa catecolaminérgica; p.Arg652Lys en el gen MHY7 (VP), que produce miocardiopatía hipertrófica, y p.Lys834Argfs*3 en DSG2 (VPP), que produce miocardiopatía arritmogénica. Asimismo se considera posible cosegregación (solo 1 familiar afecto con la variante) en los casos de p.Arg22Gln en KCNH2 (VPP), que produciría QT largo, y p.Arg439Cys en LMNA (VPP), que produciría miocardiopatía dilatada/hipertrófica, así como los casos de p.Ala688Thr en FLNC (VSI), que produciría miocardiopatía hipertrófica, y p.Tyr57His en NEBL (VSI), que produce miocardiopatía hipertrófica (hipertrofia ventricular idiopática por autopsia), hallados en 2 familiares afectos en cada caso, pero no solo 2 generaciones.
DISCUSIÓNLa MS es una de las principales causas de muerte en países industrializados, con especial relevancia cuando ocurre a personas jóvenes, y precisa un abordaje multidisciplinario debido a las causas genéticas subyacentes5. En nuestro estudio, más de la mitad de los casos tuvieron causa cardiaca y, en concreto, más de un 20% por miocardiopatía. Es conocido que hasta un 40% de las MS de jóvenes son inexplicadas (SUDS) pese a un estudio histopatológico y toxicológico apropiado, y en estos casos es obligatorio excluir una cardiopatía arritmogénica primaria subyacente debido a que puede haber familiares en riesgo6,11,12. En nuestro estudio, un 24,4% se catalogó de SUDS tras la histopatología y toxicología, y en el 70% de estos se encontró alguna variante genética.
La NGS se presenta como una herramienta muy útil en el estudio de casos de MS, y sobre todo para el posterior estudio familiar13,21. Esta nueva herramienta ha sustituido a la autopsia molecular «tradicional», utilizada previamente, que se centraba en unos pocos genes: KCNQ1, KCNH2, SCN5A y RyR214,22, mediante secuenciación Sanger, en la que el rendimiento diagnóstico estaba en torno al 0-35%23,24. En la autopsia molecular con NGS en familias con SUDS de 1-35 años, la tasa de éxito en identificar VP o VPP está en torno al 30-35%15,16,25. Pensamos que hoy en día los exomas no aportan ventajas sobre los paneles amplios de NGS, debido a la posibilidad de falsos negativos y falsos positivos (más VSI, en torno a 13/caso, y de difícil interpretación).
Se presenta un estudio prospectivo de autopsia exhaustiva molecular en casos de MS de jóvenes para demostrar su rentabilidad diagnóstica. Se consiguió ácido desoxirribonucleico (ADN) en todos los casos en que se indicó estudio genético, al tratarse de un estudio diseñado a tal efecto; el ADN se extrajo de tejido miocárdico en los casos con sangre no apta o ADN extraído de baja calidad, a diferencia de estudios previos25. Al incluirse todos los casos consecutivos ocurridos en nuestra comunidad y centralizados en un único Instituto de Medicina Legal, no hay casos perdidos como también ocurre en otros estudios. Se utilizó secuenciación masiva por NGS mediante paneles amplios de entre 194 y 380 genes relacionados con MS arrítmica, muy por encima de otros estudios similares. Se ha demostrado que con estudios con paneles amplios de genes aumenta hasta en un 20% la posibilidad de hallar variantes patogénicas26,27. Las variantes genéticas halladas se han clasificado siguiendo las recientes directrices de la ACMG/AMP20. Nuestra rentabilidad ha sido del 64,5% si se incluyen VSI, VP y VPP, y del 27,4% si solo se consideran VP y VPP. Lo mismo ocurre con los casos de SUDS, la rentabilidad considerando VSI, VP y VPP fue del 70%, cifra que sería superior que en los demás estudios, aunque si se excluye las VSI, se reduce hasta el 26,6%. El posterior estudio familiar ha permitido demostrar en varios casos la cosegregación de estas VSI o VPP con el fenotipo, con lo que ha mejorado la rentabilidad diagnóstica general al 30,6%, que sin duda seguirá aumentando tras más años de estudio y seguimiento de familiares, dado el alto porcentaje de estos con fenotipo negativo en el momento del estudio, normalmente por ser más joven que el fallecido. Hay que ser prudentes a la hora de interpretar las variantes identificadas en la autopsia molecular, y siempre hay que hacerlo dentro del marco del estudio clínico y familiar27.
La incidencia en las Islas Baleares de MS de personas de edad ≤ 50 años se sitúa en 5,8 casos/100.000 habitantes/año, superior a la mayoría de las series previas, en parte influida por la alta tasa de turistas que visitan las Islas Baleares. Si se excluye a los fallecidos desplazados, la incidencia baja a 4,2 casos/100.000 habitantes/año, cifra que no obstante sigue siendo mayor que en otras series (que además no mencionan si se incluye a pacientes desplazados)6,11,19. No hemos identificado en nuestra serie una alta frecuencia de alguna mutación fundadora que justificase esa mayor incidencia. Sí pensamos que podría tener también cierta influencia el hecho de que más del 50% de las MS tenían resultados toxicológicos positivos, si bien solo en el 10,6% fue la causa fundamental de la muerte (principalmente en relación con la cocaína). Quizá en algunos casos el tóxico, ya sea alcohol, droga ilícita o fármaco sicotrópico, podría haber actuado como factor precipitante de la muerte en el seno de una enfermedad de base.
En referencia a la edad de la población estudiada, no hay un punto de corte uniforme en todos los estudios publicados; algunos señalan en 40 años la edad para definir la «MS de joven»28. Nosotros elegimos 50 años porque pensamos que, aunque aumentaría mucho la incidencia de cardiopatía isquémica, merecían estudio las MS de esta franja de edad para que no quedasen sin diagnosticar casos hereditarios, como así ha sido: entre los 40 y los 50 años, se detectaron 25 casos de cardiopatías potencialmente hereditarias: 2 miocardiopatías hipertróficas, 5 hipertrofias ventriculares idiopáticas, 1 miocardiopatía arritmogénica, 1 miocardiopatía dilatada, 1 síndrome aórtico y 15 SUDS. En los casos de SUDS se han hallado VSI, VP o VPP en 9 (60%) y VP/VPP en 6 (40%) (3 relacionadas con miocardiopatías, 2 síndromes de QT largo y 1 taquicardia ventricular polimorfa catecolaminérgica). Por lo tanto, pensamos que la autopsia molecular debe hacerse extensiva también a esta franja de edad, dado que el número de familias que pueden beneficiarse es considerable.
En la tabla 2 se presentan los datos comparativos de las diferentes series similares publicadas6,11,12,19,25–27,29–34.
Series publicadas sobre MS. Datos comparativos
| Referencia | Años | Población | Intervalo de edades | Tipo de estudio | Incidencia/100.000 habitantes/año | N total | Causa cardiaca, % | cardiopatía isquémica, % | Miocardiopatías, % | SUDS, % | N.o de genes estudiados | Técnica de secuenciación | Estudio familiar | Rendimiento diagnóstico general |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lahrouchi et al.29 | 1985-2015 | Nueva Zelanda, Dinamarca, Reino Unido, Países Bajos | 1 a 64 | Retrospectivo | 302 | 77 | NGS | Sí | 39% | |||||
| Eckart el al.12 | 1998-2008 | Estados Unidos | ≥ 18 | Retrospectivo | 902 | 79,3 | 56,6 | 20,7 | No genética | No | ||||
| Tester et al.30 | 1998-2010 | Estados Unidos | 1 a 69 | Prospectivo | 173 | 6 | Sanger | No | ||||||
| Winkel et al.19 | 2000-2006 | Dinamarca | 1 a 35 | Retrospectivo | 2,8 | 469 | 67 | 13 | 29 | No genética | No | |||
| Margey et al.11 | 2005-2007 | Irlanda | 15-35 | Retrospectivo | 2,85 | 116 | 20,7 | 26,7 | No genética | No | ||||
| Jiménez-Jáimez et al.31 | No se especifica | Granada, España | No se especifica | Retrospectivo | 30* | 50 | 4-242 | Sanger o NGS | Sí | 33,30% | ||||
| Hertz et al.32 | 2009-2010 | Dinamarca | 0-50 | Prospectivo | 15 | 34 | NGS | No | ||||||
| Bagnall et al.6 | 2010-2012 | Australia y Nueva Zelanda | 1 a 35 | Prospectivo | 1,3 | 490 | 80 | 24 | 16 | 40 | 59-131 | NGS | Sí | 27,00% |
| Sánchez et al.26 | 2012-2016 | Cataluña, España | 0 a 50 | Prospectivo | 789 | 52,65 | 55 | NGS | No | 40,40% | ||||
| Hellenthal et al.33 | 2013 | Alemania | 1 a 40 | Prospectivo | 60 | 20 | 17 | 174 | NGS | Sí | 30% | |||
| Scheiper et al.27 | 2014-2015 | Alemania | 1 a 45 | Retrospectivo | 99 | 11 | 96 | NGS | No | |||||
| Torkamani et al.34 | 2014-2015 | Estados Unidos | 0-45 | Prospectivo | 25 | exoma | NGS | Sí | 40% | |||||
| Nunn et al.25 | No se especifica | Reino Unido, España, Dinamarca | 1 a 55 | Retrospectivo | 59 | 135 | NGS | Sí | 29% | |||||
| Ripoll-Vera et al. (presente estudio) | 2015-2019 | Islas Baleares, España | 0 a 50 | Prospectivo | 5,8 | 123 | 52,8 | 38 | 27 | 24,4 | 194-380 | NGS | Sí | 30,60% |
MS: muerte súbita; NGS: estudio genético por secuenciación masiva; SUDS: síndrome de muerte súbita inexplicada.
En los SIDS se pueden identificar variantes de canales hasta en un 10% y trabajos recientes con NGS apuntan a también un 4-7% de variantes sarcoméricas16,17. En nuestra serie de 5 casos hallamos alguna variante en 3, todas VSI, 2 en relación a canales y 1 a miocardiopatía.
El estudio familiar ha permitido además de demostrar la cosegregación de algunas variantes, detectar familiares afectos o portadores que precisan seguimiento. De hecho, se han implantado ya 5 desfibriladores como prevención primaria en familiares afectos con la misma enfermedad que el probando y estratificación de riesgo desfavorable, y 1 de ellos ya ha tenido una descarga apropiada.
CONCLUSIONESLa autopsia molecular y el estudio familiar son cruciales para avanzar en el conocimiento de la etiología de la MS de personas jóvenes. Permite diagnosticar la causa final genética de casos que con una autopsia judicial o un examen histopatológico sin genética no podría diagnosticarse, especialmente en los casos de MS inexplicada. Las nuevas técnicas de secuenciación masiva permiten mejorar el rendimiento diagnóstico, si bien el hallazgo de VSI en una alta proporción de casos supone un reto diagnóstico para el futuro. Así se puede estudiar mejor a las familias de estos pacientes, que pueden recibir un mejor consejo genético, y se podrán utilizar herramientas de prevención de riesgo más adecuadas. Creemos que es obligado formar equipos multidisciplinarios regionales que aseguren el adecuado estudio histopatológico, toxicológico, genético y familiar de los casos de MS de personas jóvenes.
FINANCIACIÓNInstituto de Investigación Sanitaria de Baleares (IdISBa), Palma de Mallorca, España.
CONFLICTO DE INTERESESNinguno.
- –
La MS de jóvenes se debe habitualmente a cardiopatías genéticas.
- –
La hallazgos en la autopsia guiarán la evaluación de los familiares de primer grado hacia descartar una cardiopatía estructural o una cardiopatía arritmogénica primaria.
- –
Los resultados genéticos en casos de MS deben ser evaluados en el contexto de las circunstancias del fallecimiento, la historia personal y familiar y los hallazgos de la autopsia. Toda esta información es esencial a la hora de interpretar las variantes genéticas y establecer el riesgo de los familiares de primer grado.
- –
Este es el primer estudio prospectivo publicado de una serie española de casos consecutivos de MS de personas jóvenes que combina autopsia forense-molecular mediante NGS utilizando paneles muy amplios de genes y estudio familiar en cascada.
- –
La rentabilidad de la autopsia molecular es elevada si se realiza un estudio exhaustivo protocolizado y se complementa con el estudio familiar, pues se llega al diagnóstico causal en la mayoría de los casos de MS de jóvenes y se podría prevenir nuevas MS entre los familiares.
- –
En el 30,6% de los SUDS se encontró alguna variante genética potencialmente relacionada con cardiopatía arritmogénica.