Palabras clave
La presente investigación ha sido financiada por becas de la Fundação Zerbini y FAPESP
INTRODUCCION
Numerosos estudios han tratado de asociar los agentes infecciosos en el desarrollo de la aterosclerosis y el infarto agudo de miocardio1,2, principalmente Chlamydia pneumoniae3-6. Sin embargo, los resultados son controvertidos7,8 y los datos serológicos no pueden discriminar a pacientes que desarrollarán un infarto agudo de miocardio (IAM)9. Se acepta que en la placa aterosclerótica la inflamación se relaciona con la inestabilidad de la placa10-12. Los factores de riesgo tradicionales sólo se asocian en algunos de los pacientes que experimentan infarto de miocardio. Un valor sanguíneo elevado de colesterol parece ser el hallazgo asociado más frecuente, y lo que resulta difícil de explicar es la razón de que el colesterol induzca inflamación. Por otra parte, la progresión acelerada de aterosclerosis en algunas enfermedades reumáticas autoinmunes, como el lupus eritematoso sistémico, el síndrome antifosfolipídico, la artritis reumatoide y la vasculitis, ha dado lugar a que los autores atribuyan el desarrollo de aterosclerosis a mecanismos autoinmunes13.
Por consiguiente, llevamos a cabo una serie de estudios analizando en profundidad las características histopatológicas de placas que experimentaron rotura y las comparamos con placas estables del mismo o diferentes pacientes, con el objetivo de estudiar los componentes y el tamaño de la placa, el remodelado de la pared y de la placa y la inflamación de la adventicia. También investigamos la presencia de algunos agentes infecciosos, utilizando diferentes técnicas (inmunohistoquímica, hibridación in situ y microscopia electrónica) con el objetivo de confirmar la fiabilidad de los resultados.
Rotura y remodelado de la placa
Diversas pruebas clínicas ponen de manifiesto que el IAM frecuentemente tiene lugar en puntos con un grado de estenosis coronaria de leve a moderada. El grado de estenosis luminal no sólo depende del depósito de placa sino también del tipo y el grado de remodelado de los vasos14,15. La angiografía coronaria no puede evaluar la cantidad de placa y el remodelado de los vasos. Algunos estudios recientes llevados a cabo con ecografía intravascular han documentado diferentes tipos de remodelado vascular en placas de composición y presentación clínica diversas16-18. Estos estudios han sugerido que un remodelado positivo se asocia con placas vulnerables, y un remodelado negativo con placas estables. En el presente estudio post mortem, probamos cuantitativamente la hipótesis de que el tamaño del ateroma coronario y el tipo de remodelado distinguen una lesión responsable de IAM fatal de una lesión equiestenótica en el mismo árbol coronario.
Se estudiaron histológicamente las principales ramas coronarias de 36 pacientes consecutivos con IAM fatal. Se compararon la lesión (grupo A) responsable de oclusión vascular e IAM fatal, y una placa estable equiestenótica (determinada histológicamente) (grupo B), obtenida en otra rama coronaria del mismo paciente. Las determinaciones morfométricas incluyeron áreas (tabla 1) de la luz, el remodelado de la placa, del vaso y el vascular, determinado mediante el área transversal relativa del vaso (área de lesión del vaso/área de referencia del vaso) × 100%. El remodelado positivo se definió como un área relativa del vaso > 105%, y el remodelado negativo como un área relativa del vaso < 95%. La determinación de la composición de la placa incluyó las áreas porcentuales ocupadas por fibrosis, lípidos, macrófagos (CD68) y células musculares lisas (HHF 35). Comparado con el grupo B, en el grupo A se identificaron mayores áreas de placa (media ± DE): 9,6 ± 1,5 frente a 4,7 ± 2,3 mm², vaso 12,7 ± 4,9 frente a 7,4 ± 3 mm² y luz 1,7 ± 1,5 frente a 1,2 ± 0,86 mm² (p < 0,01). El remodelado positivo fue más frecuente en el grupo A que en el grupo B: 21/30 (70%) frente a 8/26 (31%). El área de la placa se correlacionó positivamente con el área porcentual de lípidos (r = 0,68; p < 0,01) y con el área porcentual de macrófagos (r = 0,41; p < 0,01). El área de la placa se relacionó negativamente con el área porcentual de fibrosis (r = 0,64; p < 0,01) y con el área porcentual de células musculares lisas (r = 0,48; p < 0,01). El remodelado negativo estuvo presente en el 19% del grupo B y solamente en el 3% del grupo A (figs. 1A y B).


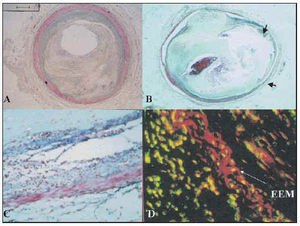
Fig. 1. A) Ejemplo de rotura de una placa trombosada de un paciente que falleció debido a un infarto agudo de miocardio, en la que se detectó un remodelado positivo asociado con un adelgazamiento de la media y una menor fibrosis adventicia. B) Correspondiente placa estable equiestenótica, en la que se identifica una capa media preservada y un menor remodelado positivo. C) Importante inflamación presente en la base de la placa, la capa media y la adventicia. D) Las imágenes confocales destacan la destrucción de la membrana elástica externa asociada con células inflamatorias.
En este estudio concluimos que las placas ateroscleróticas que inducen trombosis e IAM fatal se remodelan positivamente de forma más frecuente y tienen tendencia a ser de mayor tamaño que las placas estables.
Al analizar 34 de los 36 segmentos de la arteria coronaria (SAC) examinados en el estudio descrito previamente, valoramos si la inflamación de la adventicia se asociaría con un remodelado positivo, una menor fibrosis de la adventicia y la atenuación de la capa media en placas vulnerables. Como se ha descrito previamente, la lesión responsable (grupo A) y un segmento equiestenótico no responsable (grupo B) obtenido en las determinaciones de otra rama coronaria del mismo paciente se compararon de acuerdo con el número medio de linfocitos presentes en la adventicia y dentro de las placas. También llevamos a cabo un recuento de los microvasos adventicios, determinamos la fibrosis adventicia y, utilizando microscopia láser confocal, analizamos la membrana elástica externa19,20. Obtuvimos los resultados siguientes (tabla 2): en la adventicia, las cantidades de linfocitos y microvasos/mm² fueron, respectivamente, de 69,5 ± 88,3 y 60,9 ± 32,1 en las lesiones resposables, y de 16,4 ± 21,1 y 43,3 ± 16,1 en las lesiones estables (linfocitos p < 0,01; microvasos, p = 0,04). Los linfocitos más numerosos en la adventicia fueron los linfocitos B CD20. Dentro de las placas, el número medio de linfocitos totales fue de 24,0 ± 40,8 en las lesiones responsables y de 10,9 ± 13,2 en las estables (p = 0,17) y se identificó una diferencia significativa en los linfocitos T CD4: 6,2 ± 7,0 frente a 3,4 ± 4,1 (p < 0,05); los linfocitos más numerosos presentes en la placa fueron linfocitos T CD8. El área porcentual media de fibrosis adventicia del área transversal del vaso fue significativamente menor en las placas inestables (16,24 ± 5,07 frente a 28,95 ± 9,76%, respectivamente; p < 0,001) (figs. 1C y D). Estos hallazgos sugieren convincentemente que la inflamación crónica es responsable de la destrucción de la capa media y el colágeno de la adventicia, contribuyendo a un remodelado positivo.

Podemos establecer las siguientes conclusiones: las placas inestables manifiestan hallazgos de panarteritis crónica, acompañada de un engrosamiento, así como de un adelgazamiento de la media, y menos fibrosis que en las lesiones estables equiestenóticas. La inflamación de la adventicia contribuiría de manera importante a la inestabilidad del ateroma.
Relación entre Chlamydia pneumoniae y la rotura de la placa
A pesar del número cada vez mayor de artículos que tratan de clarificar si C. pneumoniae participa en la patogenia de la rotura de la placa y el IAM, sigue siendo un aspecto no dilucidado. La mayoría de los artículos ya publicados no cuantifican la cantidad de bacterias presentes en las lesiones (sólo si están presentes o no) y tampoco incluyen placas trombosadas que han experimentado rotura21-26. Como hemos demostrado, las placas vulnerables son las placas asociadas en realidad con la inflamación y el remodelado positivo y, por consiguiente, es preciso incluirlas. En esta parte de la presente revisión, describiremos nuestros hallazgos recientes sobre este tema, demostrando que las placas vulnerables presentan un elevado número de células infectadas por C. pneumoniae presente en la placa y en la adventicia.
En una comunicación breve previa27 y en un artículo no publicado, estudiamos los SAC trombosados responsables de IAM fatal y los comparamos con placas estables y SAC no ateroscleróticos. En todos los casos se identificó la presencia de C. pneumoniae en la adventicia, con independencia de la presencia de aterosclerosis, aunque en mayor cantidad en los segmentos de placa trombosados que habían experimentado rotura.
Se analizaron 68 SAC de pacientes fallecidos en el Heart Institute (InCor) de la University of São Paulo Medical School. La búsqueda de células positivas para C. pneumoniae se llevó a cabo utilizando una tinción de Macchiavello, inmunohistoquímica, hibridación in situ, microscopia electrónica y microscopia confocal. Se llevó a cabo un recuento de células positivas para C. pneumoniae en portas inmunoteñidos. Los SAC se dividieron en 4 grupos (tabla 3): grupo A (placa que experimentó rotura fatal a partir de 23 pacientes con IAM); grupo B (23 placas equiestenóticas que no experimentaron rotura a partir de pacientes del grupo A); grupo C (placa ateromatosa estable a partir de 11 pacientes sin IAM), y grupo D (segmentos no ateroscleróticos de la arteria coronaria a partir de 11 pacientes).

Mediante análisis de los portas inmunoteñidos, se obtuvo el número medio de células positivas para C. pneumoniae/400 × campo. Como controles positivos se utilizaron un segmento de placa que experimentó rotura, en la que se detectó un número elevado de células positivas para C. pneumoniae en el examen con microscopio electrónico, y un bloque de parafina de pulmón de conejo infectado por C. pneumoniae. Se obtuvieron los siguientes resultados: en la adventicia y en la íntima de casi todos los grupos estuvieron presentes cuerpos positivos para C. pneumoniae, detectándose mediante inmunohistoquímica, inmunofluorescencia (imágenes de láser confocal), hibridación in situ y microscopia electrónica. La cuantificación se llevó a cabo en portas teñidos con inmunohistoquímica (fosfatasa alcalina) y puso de manifiesto que el número medio de células positivas para C. pneumoniae fue significativamente mayor en el grupo A (figs. 2 y 3). Entre los grupos B, C y D no se identificaron diferencias en la cantidad de células positivas para C. pneumoniae.

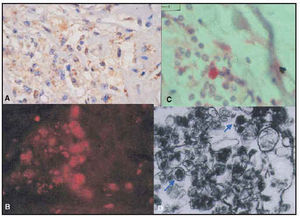
Fig. 2. A) Tejido de granulación en una placa vulnerable que contenía diversos fibroblastos y macrófagos densamente positivos para ADN-C. pneumoniae mediante hibridación in situ. B) Células positivas rosadas para los antígenos de C. pneumoniae en la adventicia mediante una técnica de inmunohistoquímica. C) Inmunofluorescencia frente a los antígenos de C. pneumoniae en una placa que ha experimentado rotura. D) Microscopia electrónica de un segmento vulnerable que contiene grandes cantidades de cuerpos de C. pneumoniae en la placa.

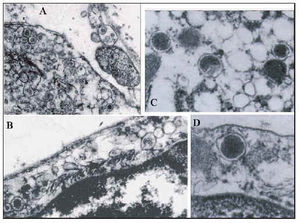
Fig. 3. Diferentes aspectos en microscopia electrónica de los cuerpos de C. pneumoniae en la célula endotelial de los microvasos de la adventicia y en los macrófagos.
Como conclusión podemos establecer que C. pneumoniae se identifica frecuentemente en la adventicia de SAC ateroscleróticos y no ateroscleróticos. Una mayor cantidad de esta bacteria se asocia con las características histopatológicas de inestabilidad de la placa. Estos hallazgos favorecen el concepto de que C. pneumoniae contribuye al desarrollo de inestabilidad de la placa.
Asociación entre Mycoplasma pneumoniae y Chlamydia pneumoniae en la placa que experimenta rotura
Después de nuestros hallazgos de que C. pneumoniae está presente con una elevada frecuencia en la adventicia de SAC y que se observa en número elevado en segmentos inestables, asociada con una inflamación aguda y un remodelado positivo, examinamos nuevos elementos que pudieran inducir la proliferación de C. pneumoniae. Revisando las muestras examinadas con microscopia electrónica, detectamos una bacteria diferente en la matriz extracelular, cerca de los cuerpos de C. pneumoniae. Esta bacteria carece de pared externa y la única bacteria que presenta dicha característica es Mycoplasma. Por consiguiente, utilizando hibridación in situ, identificamos que este microorganismo era M. pneumoniae.
Dicho hallazgo se describió en una comunicación breve: la demostración mediante microscopia electrónica e hibridación in situ de la asociación entre M. pneumoniae y C. pneumoniae en SAC trombosados28. Es bien conocido que Mycoplasma es la única bacteria que requiere colesterol en su membrana y para su proliferación29. Recientemente, se ha sugerido una posible asociación entre M. pneumoniae y la aterosclerosis, partiendo de pruebas serológicas indirectas30. Sin embargo, se ha considerado que Mycoplasma es un par&a acute;sito típico del epitelio (vías respiratorias y tracto genitourinario). Se considera que Mycoplasma no es capaz de invadir el cuerpo humano. Nuestro informe fue el primero que demostró que las paredes de los vasos son un hábitat usual de Mycoplasma.
MATERIAL Y MÉTODOS
En este estudio se utilizaron los cortes seriados de los mismos 68 SAC descritos en el estudio sobre C. pneumoniae.
También se llevó a cabo un estudio con microscopia electrónica en 4 muestras transversales adicionales.
En un caso del grupo A, se llevó a cabo una doble tinción para la detección simultánea de Chlamydia pneumoniae (mediante técnica de inmunohistoquímica) utilizando como cromógeno fosfatasa alcalina, y M. pneumoniae mediante la técnica de hibridación in situ.
La cantidad de M. pneumoniae de los casos A y B se comparó con la cantidad de células positivas para Chlamydia pneumoniae, el número de linfocitos T CD4 y CD8 y linfocitos B CD20, y el área de grasa dentro de las placas ateroscleróticas28.
Se llevaron a cabo controles negativos omitiendo la sonda o el anticuerpo primario. Como control positivo para todas las reacciones, utilizamos un tejido humano conocido previamente por ser positivo para Mycoplasma pneumoniae. Adicionalmente, para la hibridación in situ utilizamos una sonda alu repetitiva marcada con biotina (secuencia alu 1/alu 2) (Research Genetics, A1, EE.UU.) como control positivo para la técnica y una sonda plásmido ADN/biotinilada como control negativo (Dako, Carpinteria, Ca, EE.UU.).
RESULTADOS
Hibridación in situ
Se detectó C. pneumoniae como pequeños gránulos parduscos positivos en el área de grasa de las placas (fig. 4), principalmente en las inestables (grupo A).

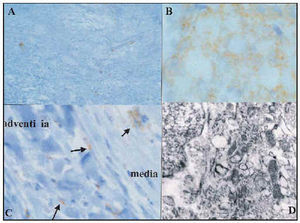
Fig. 4. A) y B) Menos M. pneumoniae entre el tejido fibroso de una placa estable. C) También se detectó M. pneumoniae en la media y la adventicia, habitualmente en las formas más extensas de estos segmentos de la arteria coronaria. D) Núcleo lipídico necrótico de una placa vulnerable en la que se detectan numerosos micoplasmas entre diversas estructuras membranosas que representarían la bacteria degenerada.
En los segmentos estables (grupos B y C), se detectó una menor cantidad de M. pneumoniae en la placa (fig. 4A) mientras que en la media prácticamente estaba ausente y en la adventicia estaba ausente o se detectó en pequeña cantidad.
En el grupo D (grupo no aterosclerótico), M. pneumoniae estaba ausente o se detectaron cantidades muy pequeñas en el espacio subendotelial.
Morfometría y comparación con el número de Chlamydia pneumoniae, número de linfocitos y porcentaje de grasa
En la tabla 4 se incluye el área porcentual de M. pneumoniae y el número medio de células positivas para C. pneumoniae (grupos A, B, C y D), el área porcentual de grasa y el número medio de linfocitos/mm² (grupos A y B).

El área porcentual ocupada por M. pneumoniae fue significativamente mayor en el grupo A (rotura de la placa) que en el grupo B (placas estables del mismo paciente) (p < 0,01). En los grupos B y C se observó una distribución similar que fue mucho mayor que en el grupo D no aterosclerótico.
Analizando las placas estables y que experimentaron rotura a partir de los grupos A y B, se obtuvo una correlación significativa (prueba de Pearson) entre el área porcentual ocupada por M. pneumoniae en la placa y el área porcentual de grasa en la placa (r = 0,69). Se identificó una importante correlación con el número medio de linfocitos T CD4+ (r = 0,37), linfocitos T CD8+ (r = 0,37) y linfocitos B CD20+ (r = 0,018).
Microscopia electrónica
Confirmando los resultados de la inmunohistoquímica y la hibridación in situ descritos previamente, se observó una gran cantidad de C. pneumoniae en los segmentos inestables en el interior de los monocitos sanguíneos, los macrófagos intersticiales, las células musculares lisas y los fibroblastos. También se detectaron otras formas de bacterias con un tamaño similar a C. pneumoniae pero diferentes desde un punto de vista morfológico (fig. 4D). Pudieron identificarse como Mycoplasma por sus características morfológicas las pequeñas estructuras redondeadas, de 0,1 a 0,4 μm de diámetro, que carecían de pared externa y contenían material granulado similar a la cromatina. Se puso de manifiesto que estaban adheridas a los vasa vasorum de las células endoteliales y, dentro de las células endoteliales, en el intersticio y en el citoplasma de los macrófagos, habitualmente cerca de los cuerpos de C. pneumoniae. Dentro de las placas de grasa, Mycoplasma se caracterizó por una forma redondeada y pequeño tamaño, entre el material ateromatoso necrótico; en la capa media y la adventicia habitualmente se detectaron en formas más extensas, cilíndricas, que contenían un mayor número de gránulos densos al microscopio electrónico recubiertos de una envoltura de membrana exclusiva.
En los segmentos de placa estable se identificó una menor cantidad de M. pneumoniae, y en la adventicia se detectaron formas mayores con gránulos densos al microscopio electrónico.
DISCUSION
Es bien conocido que Mycoplasma es el microorganismo más pequeño caracterizado por autorreproducción14. Para sobrevivir, requiere colesterol ya que su membrana externa está constituida por esta sustancia, una propiedad exclusiva entre las procariotas14. Se ha descrito una asociación con C. pneumoniae en infecciones respiratorias31,32. Se ha considerado un parásito de las superficies epiteliales.
En el presente estudio, identificamos que M. pneumoniae está presente en todas las áreas de grasa de ateroma, principalmente en la placa vulnerable, ya que suelen ser más ricas en colesterol. También detectamos mayores cantidades de M. pneumoniae en asociación con la inflamación intraplaca elevada, principalmente debido a linfocitos T CD8. Es bien conocido que M. pneumoniae induce una ligera respuesta inmune constituida básicamente por linfocitos T33. M. pneumoniae también explicaría el elevado número de células musculares lisas apoptósicas y macrófagos espumosos de las placas inestables34, ya que los micoplasmas son capaces de oxidar la membrana célular del huésped induciendo su apoptosis35. Adicionalmente, es bien conocido que los micoplasmas dan lugar a un aumento de la producción de citocinas por las células inflamatorias36, lo que explicaría la mayor concentración de citocinas observada habitualmente en las placas vulnerables37.
En las placas inestables se observó una asociación signficativa entre un número elevado de células positivas para C. pneumoniae y una cantidad elevada de M. pneumoniae (p < 0,05).
Mycoplasmas y Chlamydias degenerados explicarían el mayor número de componentes de membrana observado en los ateromas38,39.
Algunos estudios han demostrado que Mycoplasma induce una mayor virulencia de otros microorganismos40. Utilizando técnicas de doble tinción (reacción inmunohistoquímica frente a C. pneumoniae e hibridación in situ frente a M. pneumoniae) confirmamos el hallazgo de microscopia electrónica de células coinfectadas. Recientemente, ciertos hallazgos serológicos han indicado una correlación entre anticuerpos frente a M. pneumoniae y la aterosclerosis coronaria15.
Se ha sugerido que Mycoplasma participa en un elevado número de enfermedades humanas, como las neumonías, la artritis y la uretritis41, la progresión del sida, el síndrome de fatiga crónica, síndrome del distrés respiratorio agudo, etc.42. Estas ideas proceden de los hallazgos serológicos o del cultivo en algunos de los casos publicados.
No pudimos determinar cómo alcanzan el espacio subendotelial. ¿Son capaces de inducir una disfunción endotelial que favorecería la entrada de LDL en la placa? ¿Estarían presentes en la pared de la íntima y proliferarían en presencia de colesterol? Algunos aspectos que detectamos con microscopia electrónica, como las pequeñas estructuras similares a Mycoplasma en íntimo contacto con las células endoteliales, favorecerían el concepto de que alcanzan el espacio subendotelial a partir de la sangre. Mycoplasma posee lipoproteínas ancladas en la cara externa de su membrana plasmática, lo que se supone que es un mecanismo de evasión eficaz a partir del sistema de defensa inmune del huésped43.
En conclusión, describimos una serie de estudios que favorecen convincentemente el concepto de que la aterosclerosis y la rotura de la placa son complicaciones asociadas con agentes infecciosos. M. pneumoniae está presente en casi todos los ateromas coronarios. Su asociación con C. pneumoniae explicaría la mayor cantidad de C. pneumoniae, la inflamación y la inestabilidad de la placa. La gran cantidad de C. pneumoniae presente en los fibroblastos de la adventicia y las células musculares lisas es compatible con la hipótesis de que esta bacteria también favorecería el remodelado positivo del vaso9.
También especulamos que las células musculares lisas, los fibroblastos y la fibrosis que forman el ateroma podrían ser una reacción inmune del huésped, circunscribiendo los agentes infecciosos. La inflamación en la placa y la apoptosis de los componentes de la cápsula que intervienen en la patogenia de la rotura de la placa estarían influidas por la presencia de Mycoplasma.
Correspondencia: Dr. J.A.F. Ramires, MD,PhD, FESC.
Clinical Division. Heart Institute (InCor).
University of São Paulo Medical School.
Av. Dr. Eneas C. Aguiar, 44.
CEP-05403-900 São Paulo SP. Brasil.
Correo electrónico: jramires@incor.usp.br