
Las mutaciones en MYBPC3 son causa de miocardiopatía hipertrófica (MCH). A pesar de que la mayoría de ellas producen una proteína truncada, la gravedad del fenotipo es diversa. Se describe el fenotipo clínico de una nueva mutación en MYBPC3, p.Pro108Alafs*9, presente en 13 familias del sur de España, y se compara con la mutación de MYBPC3 con mayor prevalencia en dicha región (c.2308+1 G>A).
MétodosSe estudió a 107 familiares de 13 casos índice que tenían diagnóstico de MCH y portaban la mutación p.Pro108Alafs*9. Se realizó un análisis del árbol genealógico, junto con una evaluación clínica y determinación del genotipo.
ResultadosSe identificó en total a 54 portadores de la mutación p.Pro108Alafs*9, de los que 39 tenían MCH. Hubo 5 casos de muerte súbita en las 13 familias. La penetrancia de la enfermedad aumentaba a medida que se incrementaba la edad, y los pacientes con MCH fueron con más frecuencia varones, y estos contrajeron la enfermedad más precozmente que las mujeres. El fenotipo fue similar en la p.Pro108Alafs*9 y la c.2308+1 G>A, pero se observaron diferencias en varios factores de riesgo y en la supervivencia. Hubo tendencia a mayor masa ventricular izquierda en la p.Pro108Alafs*9 que en la c.2308+1G>A. La resonancia magnética cardiaca reveló una extensión y un patrón de fibrosis similares en ambas.
ConclusionesLa mutación p.Pro108Alafs*9 se asoció a MCH, alta penetrancia y aparición de la enfermedad a mediana edad.
Palabras clave
La miocardiopatía hipertrófica (MCH) es la cardiopatía hereditaria más frecuente, y afecta a 1 de cada 500 personas de la población general1,2. Clásicamente, se define como un ventrículo izquierdo (VI) hipertrofiado y no dilatado en ausencia de causa alguna capaz de producir la magnitud de la hipertrofia existente, como una sobrecarga de presión o enfermedades de almacenamiento o infiltrantes3,4. Pueden identificarse mutaciones genéticas en aproximadamente un 60% de los pacientes, y las más frecuentes son las que se dan en los genes que codifican proteínas del sarcómero cardiaco. Estas mutaciones se caracterizan por una penetrancia incompleta y una expresión clínica variable5. El gen afectado con más frecuencia es MYBPC3, que codifica la proteína de unión a la miosina C6,7. Hasta la fecha se han descrito más de 150 mutaciones causantes de MCH en el MYBPC3. A diferencia de otros genes que causan enfermedades, en los que la mayoría de las mutaciones son con cambio de sentido, aproximadamente un 70% de las mutaciones de MYBPC3 dan lugar a un desplazamiento de marco de lectura y crean un codón de terminación prematuro que da lugar a una proteína truncada en el extremo carboxiterminal6,8. La información existente sobre la correlación entre genotipo y fenotipo continúa siendo poco sólida, y parece que distintas mutaciones en el mismo gen (incluidas las mutaciones con el mismo efecto en la proteína) se comportan de manera diferente por lo que respecta a su forma de presentación y sus consecuencias clínicas9–11. No obstante, la evidencia existente se basa en un pequeño número de estudios y descripciones procedentes de pequeños grupos de pacientes. El objetivo principal del presente estudio es establecer la patogenicidad de estas mutaciones y describir el fenotipo clínico de una nueva mutación en MYBPC3 (p.Pro108Alafs*9) presente en 13 familias diferentes del sur de España. El objetivo secundario es comparar el fenotipo de las 2 mutaciones de MYBPC3 con mayor prevalencia descritas en España: p.Pro108Alafs*9 y c.2308+1 G>A (IVS23+1 G>A).
MÉTODOSPuede consultarse el apartado de «Métodos» ampliado en el .
Población del estudioSe incluyó en el estudio a los 13 casos índice con MCH, aparentemente no emparentados (edad, 40,7±14,6 años; 9 varones [75%]), que eran portadores de la misma nueva mutación p.Pro108Alafs*9 en el gen MYBPC3 (número de acceso de GenBank 4607). A todos los pacientes se les realizó una estratificación del riesgo y se aplicó un tratamiento basado en las guías recomendadas12. Se elaboró un árbol genealógico de cada paciente, y se examinó a los familiares de primer grado empleando el mismo protocolo. Se compararon los fenotipos correspondientes a las mutaciones p.Pro108Alafs*9 y c.2308+1 G>A.
En el apartado «Métodos» ampliado del se puede consultar una descripción detallada del estudio genético, el aislamiento del ARN, la síntesis del ADN complementario, las amplificaciones del ADN complementario de MYBPC3, el análisis con puromicina y el análisis estadístico.
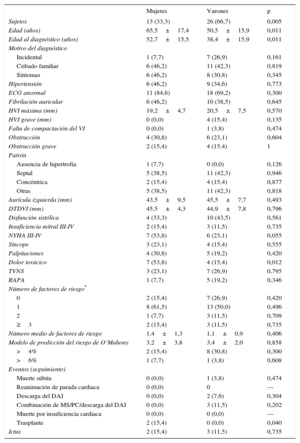
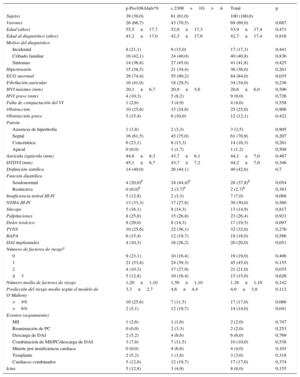
RESULTADOSTras identificar una nueva mutación en el gen MYBPC3 (p.Pro108Alafs*9), se confirmó el efecto fundador. Se evaluó en total a 107 individuos (media de edad, 42,0±20,1 años; 52 varones [48,6%]) de las 13 familias con la mutación p.Pro108Alafs*9. En el estudio clínico, 39 individuos (36,4%) cumplían los criterios diagnósticos de MCH (tabla 1), se clasificó a 7 (6,5%) como posibles casos de MCH y en 45 (42,1%) se consideró que no había afección clínica13. Se excluyó a 16 familiares con anomalías cardiacas no relacionadas.
Características de los 39 portadores de la mutación p.Pro108Alafs*9 con afección clínica
| Mujeres | Varones | p | |
|---|---|---|---|
| Sujetos | 13 (33,3) | 26 (66,7) | 0,005 |
| Edad (años) | 65,5±17,4 | 50,5±15,9 | 0,011 |
| Edad al diagnóstico (años) | 52,7±15,5 | 38,4±15,9 | 0,011 |
| Motivo del diagnóstico | |||
| Incidental | 1 (7,7) | 7 (26,9) | 0,161 |
| Cribado familiar | 6 (46,2) | 11 (42,3) | 0,819 |
| Síntomas | 6 (46,2) | 8 (30,8) | 0,345 |
| Hipertensión | 6 (46,2) | 9 (34,6) | 0,773 |
| ECG anormal | 11 (84,6) | 18 (69,2) | 0,300 |
| Fibrilación auricular | 6 (46,2) | 10 (38,5) | 0,645 |
| HVI máxima (mm) | 19,2±4,7 | 20,5±7,5 | 0,570 |
| HVI grave (mm) | 0 (0,0) | 4 (15,4) | 0,135 |
| Falta de compactación del VI | 0 (0,0) | 1 (3,8) | 0,474 |
| Obstrucción | 4 (30,8) | 6 (23,1) | 0,604 |
| Obstrucción grave | 2 (15,4) | 4 (15,4) | 1 |
| Patrón | |||
| Ausencia de hipertrofia | 1 (7,7) | 0 (0,0) | 0,126 |
| Septal | 5 (38,5) | 11 (42,3) | 0,946 |
| Concéntrica | 2 (15,4) | 4 (15,4) | 0,877 |
| Otras | 5 (38,5) | 11 (42,3) | 0,818 |
| Aurícula izquierda (mm) | 43,5±9,5 | 45,5±7,7 | 0,493 |
| DTDVI (mm) | 45,5±4,3 | 44,9±7,8 | 0,796 |
| Disfunción sistólica | 4 (33,3) | 10 (43,5) | 0,561 |
| Insuficiencia mitral III-IV | 2 (15,4) | 3 (11,5) | 0,735 |
| NYHA III-IV | 7 (53,8) | 6 (23,1) | 0,055 |
| Síncope | 3 (23,1) | 4 (15,4) | 0,555 |
| Palpitaciones | 4 (30,8) | 5 (19,2) | 0,420 |
| Dolor torácico | 7 (53,8) | 4 (15,4) | 0,012 |
| TVNS | 3 (23,1) | 7 (26,9) | 0,795 |
| RAPA | 1 (7,7) | 5 (19,2) | 0,346 |
| Número de factores de riesgo* | |||
| 0 | 2 (15,4) | 7 (26,9) | 0,420 |
| 1 | 8 (61,5) | 13 (50,0) | 0,496 |
| 2 | 1 (7,7) | 3 (11,5) | 0,709 |
| ≥3 | 2 (15,4) | 3 (11,5) | 0,735 |
| Número medio de factores de riesgo | 1,4±1,3 | 1,1±0,9 | 0,406 |
| Modelo de predicción del riesgo de O’Mahony | 3,2±3,8 | 3,4±2,0 | 0,858 |
| >4% | 2 (15,4) | 8 (30,8) | 0,300 |
| >6% | 1 (7,7) | 1 (3,8) | 0,608 |
| Eventos (seguimiento) | |||
| Muerte súbita | 0 (0,0) | 1 (3,8) | 0,474 |
| Reanimación de parada cardiaca | 0 (0,0) | 0 | — |
| Descarga del DAI | 0 (0,0) | 2 (7,6) | 0,304 |
| Combinación de MS/PC/descarga del DAI | 0 (0,0) | 3 (11,5) | 0,202 |
| Muerte por insuficiencia cardiaca | 0 (0,0) | 0 (0,0) | — |
| Trasplante | 2 (15,4) | 0 (0,0) | 0,040 |
| Ictus | 2 (15,4) | 3 (11,5) | 0,735 |
DAI: desfibrilador automático implantable; DTDVI: diámetro telediastólico del VI; ECG: electrocardiograma; HVI: grosor de la pared del VI; MS: muerte súbita; NYHA: clase de disnea de la New York Heart Association; PC: parada cardiaca; RAPA: respuesta anormal de la presión arterial durante el ejercicio; TVNS: taquicardia ventricular no sostenida en monitorización Holter; VI: ventrículo izquierdo.
HVI grave: si HVI máximo ≥ 30 mm; obstrucción: gradiente de tracto de salida del VI>30mmHg; obstrucción grave: gradiente de tracto de salida del VI ≥ 90mmHg; patrón 2: subtipo morfológico de hipertrofia según McKenna et al.13; aurícula izquierda: diámetro de la aurícula izquierda (mm); Henry (%): porcentaje del DTDVI esperado; dolor torácico: dolor torácico con el esfuerzo.
Los valores expresan n (%) o media±desviación estándar.
La mayoría de los pacientes tenían una hipertrofia moderada o grave (20,1±6,7mm) y 4 tenían una hipertrofia>30mm, todos ellos varones. Uno de los portadores presentaba unas características compatibles con falta de compactación ventricular izquierda. Trece portadores tenían síntomas limitantes (clase de disnea III-IV de la New York Heart Association [NYHA]) y 14, disfunción sistólica. El 41,0% tenía historia de fibrilación auricular.
Los pacientes con MCH eran con mayor frecuencia varones (p=0,005). Las mujeres mostraban un inicio de la enfermedad significativamente más tardío, pero tenían más síntomas (NYHA III-IV, el 53,8 frente al 23,1%; p=0,055) y sufrieron dolor torácico con más frecuencia (p=0,012) que los varones. El patrón de la hipertrofia, el electrocardiograma y el perfil de riesgo no mostraron diferencias entre los sexos.
El estudio genético de los familiares permitió identificar a 54 portadores (29 varones) y 42 no portadores, mientras que no se dispuso de muestras de ADN de 11 familiares. En total, 39 portadores (72,2%) presentaban MCH, se consideró a 2 (3,7%) como posiblemente afectados y en 13 (24,1%) se consideró que no había afección clínica.
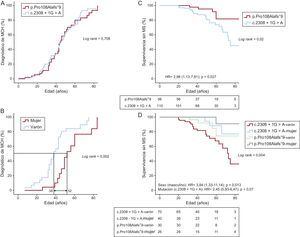
La penetrancia de la enfermedad era mayor a medida que aumentaba la edad, de tal manera que la probabilidad de que se diagnosticara la enfermedad era del 50% a la edad de 44 años (figura 1A). Los varones tendían a contraer la enfermedad antes que las mujeres (la probabilidad de sufrir MCH a los 38 años los varones y 52 las mujeres era del 50%; p=0,002) (figura 1B).
 en los portadores de 2 mutaciones: p.Pro108Alafs*9 frente a c.2308+1G>A. B: penetrancia de la enfermedad (miocardiopatía hipertrófica) en los portadores de la mutación p.Pro108Alafs*9. C: supervivencia libre de muerte súbita/descarga del desfibrilador automático implantable de los 2 grupos de portadores de diferentes mutaciones. D: supervivencia libre de muerte súbita de los portadores de 1 de las 2 mutaciones, por sexo. En A y B se incluyó a los 54 portadores de p.Pro108Alafs*9 (39 afectados, 2 posiblemente afectados y 13 no afectados) y 86 portadores de c.2308+1G>A (61 afectados y 25 no afectados). En C y D, se incluyó a los 53 portadores vivos y 3 casos de muerte súbita (2 históricos, 1 de muerte súbita) con la mutación p.Pro108Alafs*9 y 90 portadores vivos y 20 casos de muerte súbita (17 históricos, 2 de parada cardiaca reanimada, 1 de muerte súbita) con la mutación c.2308+1G>A. Dos descargas apropiadas del desfibrilador automático implantable en el primer grupo y 4 en el segundo se consideraron equivalentes de muerte súbita para los fines del análisis de la supervivencia. El número total de individuos incluidos en el análisis fue 166 («Métodos» del material suplementario). DAI: desfibrilador automático implantable; HR: hazard ratio; MCH: miocardiopatía hipertrófica; MS: muerte súbita.")
A: penetrancia de la enfermedad (miocardiopatía hipertrófica) en los portadores de 2 mutaciones: p.Pro108Alafs*9 frente a c.2308+1G>A. B: penetrancia de la enfermedad (miocardiopatía hipertrófica) en los portadores de la mutación p.Pro108Alafs*9. C: supervivencia libre de muerte súbita/descarga del desfibrilador automático implantable de los 2 grupos de portadores de diferentes mutaciones. D: supervivencia libre de muerte súbita de los portadores de 1 de las 2 mutaciones, por sexo. En A y B se incluyó a los 54 portadores de p.Pro108Alafs*9 (39 afectados, 2 posiblemente afectados y 13 no afectados) y 86 portadores de c.2308+1G>A (61 afectados y 25 no afectados). En C y D, se incluyó a los 53 portadores vivos y 3 casos de muerte súbita (2 históricos, 1 de muerte súbita) con la mutación p.Pro108Alafs*9 y 90 portadores vivos y 20 casos de muerte súbita (17 históricos, 2 de parada cardiaca reanimada, 1 de muerte súbita) con la mutación c.2308+1G>A. Dos descargas apropiadas del desfibrilador automático implantable en el primer grupo y 4 en el segundo se consideraron equivalentes de muerte súbita para los fines del análisis de la supervivencia. El número total de individuos incluidos en el análisis fue 166 («Métodos» del ). DAI: desfibrilador automático implantable; HR: hazard ratio; MCH: miocardiopatía hipertrófica; MS: muerte súbita.
Hubo un total de 5 casos de muerte súbita (MS)/descarga de un desfibrilador automático implantable (DAI) (2 históricos, 1 MS, 2 descargas del DAI apropiadas) (50±13 años; 3 varones) en las 13 familias con la mutación (3 con diagnóstico definitivo de MCH). Se realizó un seguimiento de la cohorte durante una media de 72±53 meses. El único paciente que falleció de MS durante el seguimiento fue un varón de 57 años con antecedentes familiares de MS cardiaca y 2 episodios de síncope. El paciente presentaba una hipertrofia del VI de 20mm con obstrucción y síntomas limitantes (NYHA IV). Dos mujeres precisaron trasplante cardiaco y 5 pacientes sufrieron un ictus (media de edad, 60±15 años; 3 varones).
Comparación del fenotipo causado por mutaciones de truncamiento en MYBPC3Se comparó el fenotipo de MCH en los 2 grupos de pacientes portadores de las 2 mutaciones diferentes. Ambas mutaciones modifican la longitud total de MYBPC3 (figura 2). Las características clínicas de los 39 afectados portadores de la mutación p.Pro108Alafs*9 se compararon con las de los 61 afectados portadores de la mutación c.2308+1G>A (tabla 2).

Comparación de las características clínicas de los portadores afectados de miocardiopatía hipertrófica de una de las dos mutaciones más prevalentes en nuestra región: p.Pro108Alafs*9 y c.2308+1G>A
| p.Pro108Alafs*9 | c.2308+1G>A | Total | p | |
|---|---|---|---|---|
| Sujetos | 39 (39,0) | 61 (61,0) | 100 (100,0) | |
| Varones | 26 (66,7) | 43 (70,5) | 69 (69,0) | 0,687 |
| Edad (años) | 55,5±17,7 | 52,9±17,3 | 53,9±17,4 | 0,471 |
| Edad al diagnóstico (años) | 43,2±17,0 | 42,3±17,6 | 42,7±17,4 | 0,816 |
| Motivo del diagnóstico | ||||
| Incidental | 8 (21,1) | 9 (15,0) | 17 (17,3) | 0,441 |
| Cribado familiar | 16 (42,1) | 24 (40,0) | 40 (40,8) | 0,836 |
| Síntomas | 14 (36,8) | 27 (45,0) | 41 (41,8) | 0,425 |
| Hipertensión | 15 (38,5) | 21 (34,4) | 36 (36,0) | 0,261 |
| ECG anormal | 29 (74,4) | 55 (90,2) | 84 (84,0) | 0,035 |
| Fibrilación auricular | 16 (41,0) | 18 (29,5) | 34 (34,0) | 0,236 |
| HVI máximo (mm) | 20,1±6,7 | 20,9±5,6 | 20,6±6,0 | 0,506 |
| HVI grave (mm) | 4 (10,3) | 5 (8,2) | 9 (9,0) | 0,726 |
| Falta de compactación del VI | 1 (2,6) | 3 (4,9) | 4 (4,0) | 0,558 |
| Obstrucción | 10 (25,6) | 15 (24,6) | 25 (25,0) | 0,906 |
| Obstrucción grave | 5 (15,4) | 6 (10,0) | 12 (12,1) | 0,422 |
| Patrón | ||||
| Ausencia de hipertrofia | 1 (3,8) | 2 (3,3) | 3 (3,5) | 0,905 |
| Septal | 16 (61,5) | 45 (75,0) | 61 (70,9) | 0,207 |
| Concéntrica | 6 (23,1) | 8 (13,3) | 14 (16,3) | 0,261 |
| Apical | 0 (0,0) | 1 (1,7) | 1 (1,2) | 0,508 |
| Aurícula izquierda (mm) | 44,8±8,3 | 43,7±6,1 | 44,1±7,0 | 0,467 |
| DTDVI (mm) | 45,1±6,7 | 43,7±7,2 | 44,2±7,0 | 0,346 |
| Disfunción sistólica | 14 (40,0) | 26 (44,1) | 40 (42,6) | 0,7 |
| Función diastólica | ||||
| Seudonormal | 4 (20,0)b | 24 (44,4)b | 28 (37,8)b | 0,054 |
| Restrictiva | 0 (0,0)b | 2 (3,7)b | 2 (2,7)b | 0,383 |
| Insuficiencia mitral III-IV | 5 (12,8) | 2 (3,3) | 7 (7,0) | 0,068 |
| NYHA III-IV | 13 (33,3) | 17 (27,8) | 30 (30,0) | 0,560 |
| Síncope | 5 (16,1) | 8 (14,3) | 13 (14,9) | 0,817 |
| Palpitaciones | 8 (25,8) | 15 (26,8) | 23 (26,4) | 0,921 |
| Dolor torácico | 9 (29,0) | 8 (14,3) | 17 (19,5) | 0,097 |
| TVNS | 10 (25,6) | 22 (36,1) | 32 (32,0) | 0,276 |
| RAPA | 6 (15,4) | 12 (19,7) | 18 (18,0) | 0,586 |
| DAI implantados | 4 (10,3) | 16 (26,2) | 20 (20,0) | 0,051 |
| Número de factores de riesgoa | ||||
| 0 | 9 (23,1) | 10 (16,4) | 19 (19,0) | 0,406 |
| 1 | 21 (53,8) | 24 (39,3) | 45 (45,0) | 0,155 |
| 2 | 4 (10,3) | 17 (27,9) | 21 (21,0) | 0,035 |
| ≥3 | 5 (12,8) | 10 (16,4) | 15 (15,0) | 0,626 |
| Número medio de factores de riesgo | 1,20±1,10 | 1,50±1,10 | 1,38±1,10 | 0,142 |
| Predicción del riesgo medio según el modelo de O’Mahony | 3,3±2,7 | 4,6±4,4 | 4,0±3,8 | 0,112 |
| >4% | 10 (25,6) | 7 (11,5) | 17 (17,0) | 0,066 |
| >6% | 2 (5,1) | 12 (19,7) | 14 (14,0) | 0,041 |
| Eventos (seguimiento) | ||||
| MS | 1 (2,6) | 1 (1,6) | 2 (2,0) | 0,747 |
| Reanimación de PC | 0 (0,0) | 2 (3,3) | 2 (2,0) | 0,253 |
| Descarga de DAI | 2 (5,2) | 4 (6,6) | 6 (6,0) | 0,769 |
| Combinación de MS/PC/descarga de DAI | 3 (7,8) | 7 (11,5) | 10 (10,0) | 0,538 |
| Muerte por insuficiencia cardiaca | 0 (0,0) | 4 (6,6) | 4 (4,0) | 0,103 |
| Trasplante | 2 (5,2) | 1 (1,6) | 3 (3,0) | 0,318 |
| Cardiacos combinados | 5 (12,8) | 12 (19,7) | 17 (17,0) | 0,374 |
| Ictus | 5 (12,8) | 3 (4,9) | 8 (8,0) | 0,155 |
DAI: desfibrilador automático implantable; DTDVI: diámetro telediastólico del VI; ECG: electrocardiograma; HVI: grosor de la pared del VI; MS: muerte súbita; NYHA: clase de disnea de la New York Heart Association; PC: parada cardiaca; RAPA: respuesta anormal de la presión arterial durante el ejercicio; TVNS: taquicardia ventricular no sostenida en monitorización Holter; VI: ventrículo izquierdo.
Los valores expresan n (%) o media±desviación estándar.
(0-6). Se tuvieron en cuenta los factores de riesgo para la muerte súbita: TVNS, RAPA si edad<45 años, antecedentes familiares de muerte súbita, íncope, HVI grave y gradiente grave (> 90mmHg).
Según los datos disponibles. HVI grave: si HVI máximo ≥ 30 mm; obstrucción: gradiente de tracto de salida del VI>30mmHg; obstrucción grave: gradiente de tracto de salida del VI ≥ 90mmHg; patrón 2: subtipo morfológico de hipertrofia según McKenna et al13; aurícula izquierda: diámetro de la aurícula izquierda (mm); Henry (%): porcentaje del DTDVI esperado; dolor torácico: dolor torácico con el esfuerzo; cardiacos combinados: muerte cardiaca (MS, muerte por insuficiencia cardiaca), PC, descarga de DAI y trasplante.
El fenotipo fue similar en la c.2308+1G>A y la p.Pro108Alafs*9. Sin embargo, la proporción de pacientes con un electrocardiograma característico de MCH (55 [90,2%] frente a 29 [74,4%]; p=0,035), con 2 o más factores de riesgo (27 [44,3%] frente a 9 [23,1%]; p=0,03) y con un riesgo de MS de O’Mahony en 5 años>6% (12 [19,7%] frente a 2 [5,1%]; p=0,041) fue significativamente superior entre los portadores de c.2308+1G>A que entre los portadores de p.Pro108Alafs*9. El número de episodios de MS/descarga del DAI durante el seguimiento fue similar entre los portadores de c.2308+1G>A y de p.Pro108Alafs*9 (7 [11,5%] frente a 3 [7,8%]; p=0,5).
El desarrollo de la hipertrofia en los 2 grupos de portadores fue similar a lo largo de su vida, tal como refleja la semejanza en la curva de probabilidad del diagnóstico de MCH en los 2 grupos. La penetrancia de la enfermedad aumentaba a un ritmo regular durante la vida a partir de los 20 años, siguiendo una forma sigmoidea (figura 1A). Al incluir a todos los pacientes vivos de la cohorte y los casos de MS (históricos y prospectivos, véase el apartado «Métodos»), la supervivencia en cuanto a la MS/descarga del DAI fue significativamente menor en el grupo con la mutación c.2308+1G>A que en el grupo de p.Pro108Alafs*9. El riesgo de MS/descarga del DAI fue mayor en el grupo de c.2308+1G>A que en el grupo de p.Pro108Alafs*9 (hazard ratio [HR]=2,98; intervalo de confianza del 95% [IC95%], 1,13-7,81; p=0,02) (figura 1C).
En el análisis de Kaplan-Meier, la peor supervivencia en cuanto a MS/descarga del DAI fue la de los varones portadores de la mutación c.2308+1G>A, mientras que entre los portadores de p.Pro108Alafs*9 la mejor fue la de las mujeres (figura 1D). En el análisis multivariable, el sexo masculino fue un factor predictivo de MS/descarga del DAI (HR=3,84; IC95%, 1,33-11,14; p=0,013 y hubo tendencia a mayor riesgo con la mutación c.2308+1G>A (HR=2,45; IC95%, 0,93-6,47; p=0,07). No hubo diferencias significativas en la supervivencia sin insuficiencia cardiaca (NYHA III-IV) o fibrilación auricular entre los 2 grupos ni según el sexo (datos no presentados).
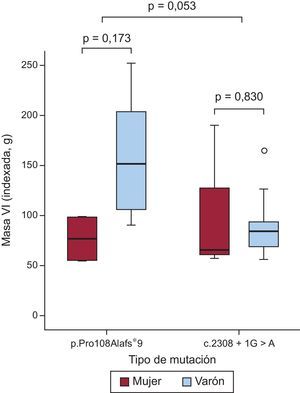
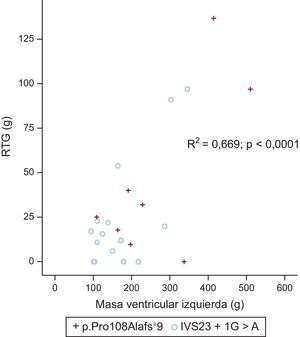
Caracterización mediante resonancia magnética cardiacaSe obtuvo una resonancia magnética cardiaca de 23 pacientes (8 con p.Pro108Alafs*9 y 15 con c.2308+1G>A) (media de edad, 44±15 años; 17 varones y 6 mujeres). Hubo tendencia a una masa ventricular izquierda mayor en el grupo de p.Pro108Alafs*9 que en el de c.2308+1G>A (269,1±138,4 frente a 173,2±80,2 g; p=0,057; p indexado=0,053) (figura 3). En 18 pacientes (78,3%) se observó realce tardío de gadolinio (RTG) (7 [87,5%] del grupo de p.Pro108Alafs*9 y 11 [73,3%] del grupo de c.2308+1G>A; p=0,9). Los patrones de RTG fueron similares en los 2 grupos. No hubo diferencias significativas entre los 2 grupos en cuanto al patrón de fibrosis. Predominaba un patrón de unión ventricular derecha-izquierda en los portadores de la mutación p.Pro108Alafs*9 (50,0%), mientras que en los portadores de la mutación c.2308+1G>A predominaba un patrón difuso septal (40,0%). No hubo diferencias en la masa con RTG ni en el porcentaje con RTG entre los dos grupos (39,9±46,8 frente a 24,6±31,3 g; p=0,3; el 12,4±11,5% frente al 13,9±11,2%; p=0,8 para los portadores de la p.Pro108Alafs*9 y la c.2308+1G>A respectivamente) (figura 4).


La presencia de la mutación p.Pro108Alafs*9 fue un factor independiente predictivo de la masa del VI (absoluta e indexada) (para la masa del VI indexada, HR=2,31; IC95%, 4,8-96,1; p=0,032). El tipo de mutación no fue un factor independiente predictivo del RTG absoluto o el porcentaje de RTG.
Sufrieron un evento arrítmico 2 pacientes (8,7%) de los que se disponía de resonancia magnética cardiaca (1 parada cardiaca reanimada y 1 descarga del DAI). Ambos pacientes eran portadores de la mutación c.2308+1G>A y tenían un patrón de RTG indicativo de fibrosis septal difusa (el 10 y el 33% de RTG de la masa total del VI).
El registro de monitorización Holter de 6 pacientes (26,0%) presentó taquicardia ventricular no sostenida; 3 de ellos (50,0%) presentaron RTG. La puntuación del riesgo de MS no se asoció a la presencia de RTG (puntuación de MS, 4,2±2,3 frente a 3,2±1,6 para la ausencia y la presencia de RTG respectivamente; p=0,3). La taquicardia ventricular no sostenida y la puntuación del riesgo de MS no se asociaron a la masa del VI ni al RTG.
Estudio histológicoEl estudio histopatológico de un corazón explantado de una mujer portadora de p.Pro108Alafs*9 mostró una hipertrofia concéntrica grave y una fibrosis intersticial difusa (). A esta paciente se le diagnosticó MCH a los 44 años. No había evidencia alguna de obstrucción. Las ecocardiografías previas al trasplante revelaron una cavidad del VI pequeña y una disfunción diastólica grave, que se consideró que era la causa de los síntomas limitantes. A pesar del tratamiento médico, la paciente precisó trasplante a los 68 años.
El estudio histológico de un varón trasplantado, portador de la mutación c.2308+1G>A, mostró grandes áreas de necrosis transmurales y heterogéneas (). Se diagnosticó MCH al paciente a los 26 años, con síntomas asociados a obstrucción. Después de 23 años de seguimiento, sufrió disfunción sistólica y recibió un trasplante a los 49 años. El examen del órgano mostró un corazón dilatado, con fibrosis extensa, desorganización e hipertrofia.
Además de ser un par de casos ilustrativos, servirían para desarrollar la hipótesis de que los portadores de una mutación causal podrían desarrollar un patrón específico de fibrosis que no se pudo demostrar en los resultados de la resonancia magnética cardiaca.
Mutaciones de truncamiento en MYBPC3Con objeto de investigar qué transcritos eran producidos por la mutación c.2308+1G>A y determinar si los transcritos truncados eran degradados por el sistema de degradación del ARN mediado por mutaciones terminadoras, se cultivaron linfocitos de portadores de las 2 mutaciones con puromicina, un inhibidor de la traducción.
La mutación c.2308+1G>A es una transición de G>A localizada en el sitio donador 3’ del intrón 23 del gen MYBPC3 que inactiva este sitio de corte y empalme. Esta mutación produce sitios de corte y empalme alternativos, que dieron lugar a 2 transcritos de mayor longitud (4.515 y 4.761 pb) que el transcrito natural (wildtype) (4.217 pb) al cultivar los linfocitos con puromicina. No obstante, solo se observó el alelo natural cuando los linfocitos se cultivaron sin puromicina, lo cual indica que los transcritos aberrantes generados por esta variante eran degradados con independencia de su longitud. Lo mismo ocurrió cuando se analizó el ARN mensajero de un portador de p.Pro108Alafs*9; en sangre, solo se expresó el alelo natural.
La mutación p.Pro108Alafs*9 es una inserción de los nucleótidos GCTGGCCCCTGCC en la posición 29 del exón 3. Esta inserción produce también un cambio en el marco de lectura: el ADN complementario aberrante da lugar a 107 residuos de aminoácidos normales de MYBPC3 y luego 8 aminoácidos nuevos, seguidos de un codón de parada prematuro en una región rica en prolina-alanina (Ala-Pro). Esto debería producir una proteína truncada muy corta de 116 aminoácidos (–91%) que carece del dominio de MYBPC3 que contiene los lugares de fosforilación y los lugares de unión de titina y miosina, en lugar de la proteína de tipo natural formada por 1.275 aminoácidos.
En la figura 2 se muestran las distintas mutaciones descritas anteriormente en MYBPC3, cada una de las cuales produce péptidos de longitudes diferentes10,11,14–17. La penetrancia varía entre el 62 y el 82%, sin que haya asociación alguna con la posición de la mutación en MYBPC3. Por el contrario, puede observarse una tendencia al aumento de la prevalencia de MS que es proporcional a la longitud del transcrito, que es del 13% para el más corto (p.Pro108Alafs*9) y máximo (67%) para el segundo más largo (IVS20-2A>G). El de mayor longitud, Q1061*, no sigue esta tendencia y la prevalencia de MS es baja.
Estudio in silicoSe estudió la patogenicidad de la mutación p.Pro108Alafs*9 empleando los criterios modificados utilizados anteriormente por Van Spaendonck-Zwarts et al.18 Una lista de las características específicas de la mutación basada en un análisis in silico realizado con el programa informático de interpretación de las mutaciones MutationTaster y la frecuencia en la población de control predijo esta variante como causa de la enfermedad con una probabilidad de 119. Fue necesario un estudio familiar para evaluar la cosegregación y finalmente para clasificar la variante como patogénica.
DISCUSIÓNFenotipo clínico de la mutación p.Pro108Alafs*9En el presente estudio se describe una nueva mutación identificada en 54 portadores de 13 familias. En total, el 72% de nuestros portadores de la mutación p.Pro108Alafs*9 tenían MCH. Nuestro estudio confirma la asociación entre la mutación p.Pro108Alafs*9 en MYBPC3 y la enfermedad con una cosegregación familiar del 100%.
Los pacientes afectados por la mutación p.Pro108Alafs*9 se caracterizan por una hipertrofia septal asimétrica con presencia de obstrucción en aproximadamente un 25% de los casos, alta penetrancia e inicio de la enfermedad a mediana edad (43±17 años). Los síntomas de insuficiencia cardiaca son predominantes, mientras que la MS es una complicación muy poco frecuente.
De modo similar a lo descrito previamente en otras mutaciones de MYBPC3, nuestros pacientes portadores mostraron una penetrancia relacionada con la edad20, con formas similares para ambas mutaciones. Tiene interés señalar que, al igual que en otras 2 series publicadas10,21, hubo un claro predominio del sexo masculino entre los portadores de p.Pro108Alafs*9 afectados, y que los varones tenían una edad inferior a la de las mujeres en el momento del diagnóstico. Sin embargo, no se han demostrado diferencias de penetrancia o del tiempo transcurrido hasta el inicio de la enfermedad entre los sexos en poblaciones amplias de pacientes con MCH15,22–24.
De manera similar a lo indicado por otras series, la mayoría de los portadores afectados se encontraban en NYHA I-II, menos del 20% refirieron síncopes y menos del 30% tenían dolor torácico25. En promedio, las mujeres se encontraban en una clase funcional de la NYHA más desfavorable y presentaban dolor torácico con mayor frecuencia. El porcentaje de pacientes con fibrilación auricular en nuestra serie (41%) fue superior al descrito por otros autores24,26.
Comparación fenotípica entre las dos mutacionesEn este estudio se compararon las características fenotípicas de las 2 mutaciones de mayor prevalencia halladas en nuestra región, p.Pro108Alafs*9 y c.2308+1G>A. Esta es una de las series más grandes publicadas con una misma mutación en MYBPC310. La mutación p.Pro108Alafs*9 fue también una mutación fundadora en nuestra cohorte pero, a diferencia de la c.2308+1G>A, produjo una tasa de episodios arrítmicos inferior en los pacientes afectados. La supervivencia sin MS/descarga del DAI (casos históricos y prospectivos) fue claramente inferior conn la mutación c.2308+1G>A que con la p.Pro108Alafs*9. Se han descrito otras mutaciones fundadoras en MYBPC3, situadas en diferentes regiones que predicen una proteína truncada en el extremo carboxiterminal debido a un codón de stop (finalización) prematuro11,12,14–17,27,28. Las mutaciones dominantes suelen causar una presión de selección negativa y tienden a desaparecer al cabo de varias generaciones. Algunas de esas mutaciones, como c.2308+1G>A, escapan a esta presión de selección y se transmiten durante generaciones debido a que la expresión de la enfermedad no se produce hasta después de alcanzada la edad reproductiva16.
Los resultados de los estudios de correlación genotipo-fenotipo en la MCH se han visto afectados por el efecto de confusión producido por el pequeño tamaño de las familias, la baja frecuencia de cada mutación causal (< 5%) y el bajo número de familias con mutaciones idénticas29. En general, las mutaciones del gen MYBPC3 se asocian a una media de edad de inicio de los síntomas más avanzada, una menor incidencia de MS y un curso clínico relativamente benigno, si bien no ha habido diferencias del fenotipo clínico atribuibles al tipo específico de mutación de MYBPC330. En consecuencia, la elevada prevalencia de estas mutaciones fundadoras brinda la oportunidad de definir sus perfiles clínicos.
Longitud del transcrito y fenotipoLa vía de degradación mediada por mutaciones con cambio de sentido es un sistema de vigilancia del ARN mensajero en el que es característico que se degraden los transcritos que contienen codones de terminación prematura, con objeto de prevenir la traducción de transcritos innecesarios o aberrantes. Un fenotipo relativamente más leve podría deberse a mutaciones con cambio de sentido que activen esta vía de degradación del ARN, con lo que reducen la expresión negativa dominante y dan lugar a una haploinsuficieia20,31,32. Se sabe que los cMYBPC truncados son degradados preferentemente por el sistema ubiquitina-proteasoma, lo cual puede deteriorar la capacidad proteolítica de dicho sistema33,34.
La longitud del transcrito de ARN mensajero podría tener un papel en el perfil de riesgo diferente para cada mutación. Se han observado diferencias importantes en las células cardiacas con MYBPC3 mutado en lo que respecta a la longitud del trascrito proteico: a) la sobrexpresión de los cMYBP-C truncados humanos en ratones transgénicos produjo unas cantidades de proteína notablemente inferiores, directamente correlacionadas con el tamaño de la proteína; b) las proteínas de mayor tamaño tienen mayor probabilidad de incorporarse al sarcómero, y c) las proteínas de menor tamaño son más propensas a bloquear el sistema de degradación ubiquitina-proteasoma, lo que da lugar a la formación de agregados de proteínas truncadas y aumento de la concentración citosólica de otras proteínas que intervienen en el crecimiento y la atrofia muscular y la apoptosis33,35. El sistema ubiquitina-proteasoma desempeña también un papel en la degradación de los receptores adrenérgicos β2 y los canales iónicos.
Tiene interés señalar que la patogenicidad de las mutaciones en titina se ha asociado recientemente con la longitud del transcrito proteico36.
En nuestros resultados se puede apreciar que la longitud del transcrito no afecta a la gravedad de la hipertrofia ni a la penetrancia de la enfermedad, pero sí al perfil de riesgo y el pronóstico de MS, que fue mucho peor en el grupo con transcritos de mayor longitud. Al analizar en detalle otras mutaciones fundadoras que truncan el gen MYBPC3, se observa que el número de eventos de MS era proporcional a la longitud del transcrito, excepto en el caso de p.Gln1061* (p=0,081) (tabla 2, «total de casos de MS/total de afectados de MCH»).
Fibrosis y riesgo de muerte súbitaEl RTG se ha asociado al perfil de riesgo de MS y la taquicardia ventricular no sostenida en la MCH26,37–39. El patrón de las uniones entre el ventrículo derecho y el VI es el más frecuente y se cree que tiene un pronóstico más benigno que el de los patrones difuso o transmural25. El RTG extenso (> 20% o>30% de la masa del VI) indica peor sustrato arrítmico39,40.
Aunque los pacientes con la mutación c.2308+1G>A de nuestro estudio presentaron una puntuación de MS significativamente peor (perfil de riesgo de MS obtenido con el modelo de O’Mahony) y una peor supervivencia libre de MS en comparación con los pacientes con la mutación p.Pro108Alafs*9, el supuesto sustrato arrítmico no se observó en el subgrupo de pacientes examinados con resonancia magnética cardiaca. El grado y el patrón de RTG fueron similares en los pacientes con la mutación p.Pro108Alafs*9 y con la mutación c.2308+1G>A. Además, y contrariamente a lo que cabría esperar, la masa del VI indexada según el sexo y la edad fue mayor en los portadores de la mutación p.Pro108Alafs*9 que en los de c.2308+1G>A (p=0,032).
CONCLUSIONESLa nueva mutación p.Pro108Alafs*9 en MYBPC3 se asocia con desarrollo de MCH a mediana edad con alta penetrancia. Predominan los síntomas de insuficiencia cardiaca, mientras que la MS es una complicación muy poco común. El riesgo de MS de los portadores de mutaciones en MYBPC3 podría estar asociado a la longitud del transcrito aberrante, pero esta hipótesis se deberá confirmar en futuros estudios.
FINANCIACIÓNEste estudio fue financiado en parte por una subvención nacional de la Sociedad Española de Cardiología-Fundación Española del Corazón (SEC-FEC/2014). Los investigadores forman parte de una red de investigación cardiovascular (RD12/0042/0049,69) y del Instituto Murciano de Investigación Biosanitaria, ambos del Instituto de Salud Carlos III-Unión Europea, Fondo Europeo de Desarrollo Regional, «Una manera de hacer Europa». M. Sabater-Molina, D. Pascual-Figal y J.R. Gimeno trabajan también en la Universidad de Murcia.
CONFLICTO DE INTERESESNinguno.
- –
La mayor parte de las mutaciones fundadoras en MYBPC3 asociadas con MCH causan una proteína truncada. No obstante, las diferencias fenotípicas y el pronóstico parecen variar según cuál sea la mutación.
- –
La guía de la Sociedad Europea de Cardiología recientemente publicada fomenta el empleo de una fórmula para estimar el riesgo de MS en la MCH. Dos variables importantes, como la información genética y la fibrosis (realce tardío de gadolinio en la resonancia magnética cardiaca) no se incluyeron en el análisis.
- –
Se presentan los resultados del análisis y la comparación de 2 cohortes amplias de pacientes portadores de 2 mutaciones de truncamiento distintas en el mismo gen (MYBPC3), que se comportan de manera diferente. Una de ellas, la mutación p.Pro108Alafs*9, es nueva y su patogenicidad está clara. La diferencia de pronóstico podría no explicarse por la gravedad de la hipertrofia o el grado de fibrosis.
- –
Los datos que se presentan aquí y los resultados de una búsqueda bibliográfica indican que habría asociación entre la longitud del transcrito y la proporción de casos de MS. Esta hipótesis concuerda con lo indicado por los experimentos celulares y los resultados de los estudios de patogenicidad de otros genes.
Queremos expresar nuestro sincero agradecimiento a las familias que aceptaron amablemente participar en el estudio.