Las hospitalizaciones por insuficiencia cardiaca crónica (ICC) continúan aumentando en Europa y Estados Unidos. Se estima que las hospitalizaciones debidas a insuficiencia cardiaca aguda descompensada (ICAD) son la causa de más del 75% de los costes de la asistencia sanitaria de los pacientes con ICC1,2. Los datos de ensayos clínicos y registros amplios han puesto de relieve que la mayor parte de las hospitalizaciones por ICAD se producen a causa de síntomas (p. ej., disnea, distensión abdominal y fatiga) y signos (como estertores pulmonares, distensión venosa yugular y edema periférico) de congestión venosa, y no por bajo gasto cardiaco3,4. Los síntomas de congestión se agravan habitualmente pocos días (3 ± 2,5 días) antes del ingreso en el hospital5. Sin embargo, estudios recientes han puesto de manifiesto que el inicio de la congestión venosa se produce mucho antes de que se manifiesten los síntomas de congestión. La determinación domiciliaria diaria del peso corporal6, la monitorización continua de las presiones intracardiacas (Chronicle, Medtronic Inc.)7 y la congestión pulmonar mediante la impedancia intratorácica (OptiVol, Medtronic Inc.)5 aportan indicios de que la congestión venosa empieza a producirse mucho antes de lo que anteriormente se había pensado en el curso de la ICAD.
La congestión venosa (caracterizada por el aumento del peso, las presiones de llenado del corazón derecho y la acumulación de líquido intratorácico) empieza a aumentar al menos 7-14 días antes de que se agraven los signos y síntomas de ICC y requieran finalmente un tratamiento intravenoso urgente5-7. Aunque el control de la congestión es un objetivo importante del tratamiento, los médicos no logran buenos resultados en lo que se refiere al tratamiento de la congestión, como pone de relieve el hecho de que un 50% de los pacientes no pierdan peso durante la hospitalización8. Este fracaso terapéutico tiene importantes consecuencias, puesto que la congestión sistémica rebelde al tratamiento es un importante factor predictivo hemodinámico del empeoramiento de la función renal, la rehospitalización y la mortalidad tras el alta de los pacientes que son hospitalizados por una ICAD9-13.
El incumplimiento de la dieta, la falta de adherencia a la medicación, el empeoramiento de las funciones sistólica o diastólica de los ventrículos izquierdo o derecho, la hipertensión, la isquemia y las arritmias son factores que pueden fomentar la retención de líquidos y la congestión venosa en los pacientes con ICC14. Sin embargo, aunque la acumulación de líquidos es más efecto que causa, una vez iniciada y mantenida tiene efectos negativos para el corazón (p. ej., fomentando la isquemia subendocárdica)15, los riñones (p. ej., reduciendo la presión de perfusión, por lo que se produce retención de sodio)12,13,16 y también, según lo indicado por nuestras observaciones iniciales, el endotelio venoso y la producción y la liberación periférica de citocinas y neurohormonas17,18.
Nuestra hipótesis es que la congestión venosa es, de por sí, un estímulo inflamatorio y hemodinámico fundamental que contribuye a la aparición y progresión de la ICAD a través de mecanismos endoteliales, neurohormonales, renales y cardiacos. El comentario que se presenta a continuación detalla la evidencia disponible que respalda esta hipótesis.
El endotelio venoso es el órgano endocrino/paracrino más grande del organismo y un regulador clave del volumen sanguíneo central, la perfusión de los órganos y la hemostasia en la ICC, a través de transiciones entre los estados latente y activado que se producen en respuesta a factores de estrés ambientales, como la distensión vascular asociada a la congestión venosa19. Con el empleo de un nuevo enfoque basado en la obtención de muestras de células endoteliales venosas y la cuantificación de la expresión de proteínas y de ARNm, hemos descrito anteriormente un aumento de los radicales libres prooxidantes y las proteínas proinflamatorias en las células del endotelio venoso obtenidas de pacientes hospitalizados por ICAD y con signos clínicos de sobrecarga de líquido20,21. Esta activación del endotelio venoso cede, en parte, con la diuresis y la mejoría clínica durante la hospitalización inicial del estudio21. Otros datos más recientes, obtenidos en animales y en seres humanos, han confirmado que la congestión venosa puede causar, por sí sola, un cambio del perfil de síntesis y endocrino del endotelio haciendo que pase de un estado latente a un estado activado con unas características prooxidantes, proinflamatorias y vasoconstrictoras que concuerdan con las observadas en pacientes con ICAD17,18. La liberación periférica resultante de neurohormonas vasoactivas y proinflamatorias (es decir, factor de necrosis tisular alfa, endotelina 1, interleucina 6 y angiotensina II) por el endotelio distendido y el tejido perivascular congestivo puede contrarrestar las adaptaciones fisiológicas que mantienen la ICC en un estado de compensación (es decir, redistribución del gasto cardiaco limitado a los órganos vitales, como riñones, corazón y cerebro). Estos procesos fisiopatológicos pueden fomentar una retención de líquidos adicional que lleve a un círculo vicioso que finalmente conducirá a que progrese una descompensación manifiesta.
En los riñones, un aumento de la congestión y la presión venosas tiene consecuencias nocivas tanto hemodinámicas como intrínsecas, puesto que reduce la perfusión de los órganos y aumenta la retención de sodio16. Es de destacar que se ha demostrado que tanto los radicales libres prooxidantes como las citocinas proinflamatorias (como el factor de necrosis tumoral alfa) liberados por el endotelio distendido en respuesta a la congestión venosa reducen la excreción renal de sodio22,23.
La congestión venosa puede tener también repercusiones negativas en la función cardiaca, pues causa isquemia subendocárdica, remodelado ventricular izquierdo, deterioro del drenaje venoso cardiaco por las venas coronarias y reducción del umbral para las arritmias15. La disminución resultante del gasto cardiaco puede causar un deterioro aún mayor de la perfusión y la función renales, con lo que se produciría una mayor retención de líquidos. Es de destacar que el endocardio cardiaco es estructuralmente idéntico al endotelio vascular y está en continuidad con él, probablemente esté activado y posiblemente contribuya a la liberación de angiotensina II que se produce en respuesta a las presiones de llenado cardiaco elevadas24.
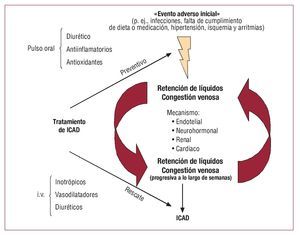
En la figura 1 se resume lo que consideramos que es una «hipótesis venocéntrica periférica unifica-dora» para explicar la fisiopatología de la ICAD. Al inicio se producen uno o varios eventos adversos de diversa etiología (p. ej., infecciones, incumplimiento de la dieta o la medicación, arritmias, isquemia o agravamiento de la hipertensión), lo que causa una retención de líquidos. La propia congestión venosa, a través de mecanismos endoteliales, neurohormonales, renales y cardiacos, puede conducir a una retención adicional de sodio y agua. La distensión vascular asociada a la congestión venosa puede cambiar el perfil de síntesis y endocrino del endotelio venoso, haciendo que pase de un estado latente a un estado activado, prooxidante, proinflamatorio y vasoconstrictor que, a su vez, fomenta la liberación periférica de neurohormonas vasoactivas y proinflamatorias. En los riñones, la congestión vascular y la activación del endotelio distendido, que entonces es ya de por sí una fuente de estrés oxidativo y de citocinas proinflamatorias, pueden causar una mayor retención de líquidos. En el corazón, las presiones de llenado elevadas producen un deterioro aún mayor de las funciones sistólica y diastólica. Posteriormente, cuando el evento o los eventos iniciales ceden, es posible que sea demasiado tarde, puesto que se ha instaurado ya el círculo vicioso que une la congestión vascular a la retención progresiva de líquidos. Los síntomas acabarán empeorando después de semanas de acumulación progresiva de líquido, lo que motivará finalmente una hospitalización por una descompensación manifiesta.

Fig. 1. Repercusiones de la congestión venosa en la fisiopatología y el tratamiento de la insuficiencia cardiaca aguda descompensada (ICAD).
La detección precoz (p. ej., mediante la monitorización continua de las presiones intracardiacas y la impedancia intratorácica) y un mejor conocimiento de la fisiopatología del ICAD pueden permitir la aplicación de futuras estrategias terapéuticas para pasar del actual «modo de rescate» de las intervenciones intravenosas tardías (con inotrópicos y diuréticos) a un «modo preventivo» de intervenciones por vía oral. Esta estrategia de tratamiento temprano puede incluir no sólo diuréticos, sino también, como cabe inferir de nuestros datos y podrá examinarse en futuros estudios, otros métodos adyuvantes como los tratamientos de corta duración (pulsos) antioxidantes o antiinflamatorios que pueden interrumpir la ICAD antes de la progresión a una descompensación manifiesta.
En resumen, la congestión venosa puede actuar como un estímulo independiente y fundamental para la aparición y la progresión de la ICAD. Es importante señalar que nuestro enfoque «venocéntrico» de la fisiopatología de la ICAD tiene como objetivo complementar los puntos de vista «cardiocéntrico», «nefrocéntrico» o «arteriocéntrico», más tradicionales, puesto que parece que todos los sistemas (es decir, el corazón, los riñones, las arterias y las venas) intervienen en los episodios que desencadenan y mantienen la ICAD.
Declaración de conflictos de intereses: Paolo C. Colombo, MD: Medtronic Inc: subvención de investigación instada por el investigador.
Full English text available from: www.revespcardiol.org
Correspondencia: Dr. P.C. Colombo.
Division of Cardiology. New York-Presbyterian Hospital.
622W 168th Street, PH 12-134. New York, NY 10032. Estados Unidos.
Correo electrónico: pcc2001@columbia.edu