La insuficiencia cardiaca con fracción de eyección reducida es un síndrome complejo en el que, como consecuencia de la disfunción del ventrículo izquierdo, se produce una hipoperfusión general que afecta a los diferentes órganos, lo que conduce a la activación de diferentes mecanismos compensatorios como el sistema renina-angiotensina-aldosterona o el sistema nervioso simpático; aunque inicialmente tratan de mantener la homeostasis del organismo, finalmente acaban siendo deletéreos. Igualmente se activan vías protectoras como la de los péptidos natriuréticos o el óxido nítrico-guanilato ciclasa. Por lo tanto, el tratamiento de la insuficiencia cardiaca debe dirigirse al reequilibrio de los diferentes sistemas neurohormonales. La reducción de la concentración de guanosina monofosfato cíclico (GMPc) se asocia con alteraciones cardiacas, vasculares y renales. El vericiguat es un estimulador oral de la guanilato ciclasa soluble que permite mejorar la vía óxido nítrico-guanilato ciclasa soluble-GMPc, lo que se traduciría en beneficios clínicos.
Palabras clave
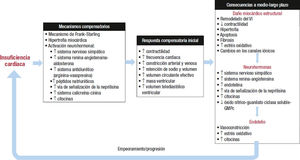
La insuficiencia cardiaca (IC) es un síndrome complejo en el que hay una alteración del llenado ventricular o de la función contráctil del corazón que acaba en la aparición de los síntomas o signos de la IC1. Como consecuencia de las disfunción del ventrículo izquierdo (VI), se produce una hipoperfusión general que afecta a los diferentes órganos y sistemas del organismo, lo que conduce a la activación de diferentes mecanismos compensatorios; aunque inicialmente tratan de mantener la homeostasis del organismo, finalmente acaban siendo
deletéreos y constituyen un auténtico círculo vicioso que se autoper- petúa, salvo que se paute el tratamiento adecuado (figura 1.)1-8.

Según la ley (o mecanismo) de Frank-Starling, cuanto mayor sea el volumen de llenado en el VI durante la diástole, mayor será el volumen sanguíneo expulsado durante la siguiente contracción sistólica, esto es, una mayor fuerza de contracción8. Sin embargo, en la IC se produce una serie de cambios estructurales que incluyen la hipertrofia ventricular, la dilatación de las cámaras cardiacas, la desorganización de los cardiomiocitos y el aumento de la tensión parietal, entre otros, que acaban causando un deterioro de la función cardiaca de tal forma que, para un mismo volumen telediastólico, el volumen telesis- tólico es menor en comparación con un corazón normal3,4,6,7.
Pero sin duda el principal mecanismo compensatorio que ocurre en el paciente con IC es la activación de los diferentes sistemas neuro-hormonales, que incluyen, principalmente, el sistema nervioso simpático, el sistema renina-angiotensina-aldosterona (SRAA), la vaso- presina, el sistema de péptidos natriuréticos (PN), la vía señalización de la neprilisina y el sistema calicreína-cinina1-8. En general, estos sistemas son mecanismos adaptativos, con un efecto beneficioso (compensatorio) inicial, como el aumento de la contractilidad cardiaca, la constricción periférica y la retención de sodio y volumen, lo que aumenta el volumen circulante efectivo, el volumen telediastó- lico y la masa ventricular2-7.
Sin embargo, lo que inicialmente son mecanismos beneficiosos se convierten en perjudiciales. Así, como consecuencia de la perpetuación de estos sistemas, se producen hipertrofia cardiaca y aumento de la masa ventricular, mayor fibrosis y remodelado ventricular que, junto con el aumento del estrés oxidativo, el estado inflamatorio y las alteraciones moleculares, finalmente producen una disminución de la capacidad contráctil del corazón1-7,9. Asimismo, en los vasos sanguíneos se produce un aumento del tono vascular, un incremento de la inflamación y una alteración de la vía óxido nítrico-guanosina monofosfato cíclico (GMPc), lo que conduce a vasoconstricción y alteración de la función endotelial. Esto se traduce en un mayor riesgo de eventos cardiovasculares y un peor pronóstico10. Además, los numerosos sistemas neurohormonales siguen interviniendo, bien por activación de vías que acaban siendo deletéreas como el sistema nervioso simpático o el SRAA, bien por la inhibición de vías protectoras cardiovasculares, como la de los PN, mediante el aumento de la señalización de la neprilisina o la inhibición del sistema óxido nítrico-guanilato ciclasa soluble-GMPc, lo que no hace sino empeorar la situación de la IC, y esto a su vez vuelve a tener un efecto estimulador de los diferentes sistemas neurohormonales, lo que completa el círculo vicioso que supone la etiopatogenia de la IC (figura 1 )1-8.
Sistemas neurohormonales implicados en la etiopatogenia de la insuficiencia cardiacaLa IC se debe entender como un síndrome con una etiopatogenia multifactorial en la que intervienen diferentes sistemas neurohormonales (algunos por activación y otros por inhibición), cada uno con sus particularidades, pero que se encuentran relacionados y se potencian entre sí. A continuación se resumen los aspectos más relevantes de cada uno de ellos.
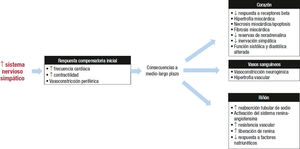
Sistema nervioso simpáticoLa reducción de la presión arterial media que ocurre en la IC, detectada tanto por los barorreceptores de los senos carotídeos y del arco aórtico como por los mecanorreceptores de la circulación car- diopulmonar, estimula la actividad simpática y, por ende, disminuye la parasimpática. Como resultado del aumento de las catecolaminas en sangre, se produce un incremento de la frecuencia y la contractilidad cardiacas que, junto con la vasoconstricción periférica, aumenta la resistencia total periférica y con ello la presión arterial media. Sin embargo, la activación persistente del sistema nervioso simpático llega a tener efectos perjudiciales en diferentes localizaciones. Así, la estimulación crónica de los miocitos por la noradrena- lina promueve su crecimiento e hipertrofia y el remodelado del mio cardio con fenómenos de necrosis celular y fibrosis, disminución de la respuesta de los receptores beta y, aunque inicialmente la nor- adrenalina está incrementada, conforme progresa la IC comienza a disminuir al reducirse las reservas, tanto por un fenómeno de agota-miento como por una disminución de su síntesis. Todos estos fenómenos producen una alteración de la función sistólica y diastólica del corazón. En el lecho vascular, la activación del sistema nervioso simpático favorece la vasoconstricción, la hipertrofia vascular, la reducción del óxido nítrico y el aumento de la inflamación. En los riñones, la estimulación del sistema nervioso simpático aumenta la reabsorción de sodio y las resistencias vasculares y favorece la activación del SRAA (figura 2.)2,5,11. En este contexto, los bloqueadores beta (BB) reducen la frecuencia cardiaca, aumentan la duración de la diástole, mejoran el llenado miocárdico y reducen la demanda de oxígeno, lo que en último término se traduce en una mejoría de la función cardiaca1.
Sistema renina-angiotensina-aldosterona
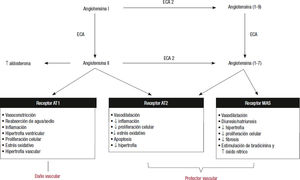
En la IC se produce una estimulación del SRAA como consecuencia de la activación de los receptores ß1 y α1 debido a la hipoperfusión renal y una reducción de la entrega de sodio a la mácula densa del túbulo distal. Esta activación del SRAA produce un aumento de la angiotensina II, que es un potente vasoconstrictor, que además estimula la activación del sistema nervioso simpático, aumenta la absorción de sodio y cloro de la zona ascendente del asa de Henle y estimula la liberación de aldosterona de la corteza adrenal, la vasoconstricción arteriolar tanto sistémica como renal y la liberación de la vasopresina de la hipófisis. En resumen, mediante la activación del SRAA inicialmente se consigue mantener el volumen intravascular y retener sodio, lo que permite mantener la presión arterial y mejorar la perfusión tisular. Sin embargo, esta activación mantenida produce efectos deletéreos (hipertrofia ventricular y vascular, fibrosis, rigidez arterial, aumento del estrés oxidativo y del estado inflamatorio, vasoconstricción periférica, estimulación del sistema nervioso simpático, disfunción endotelial, disfunción de los barorreceptores, etc.) que aumentan la poscarga persistentemente y promueven el fallo miocár- dico (figura 3.)2,12-14. Tanto los inhibidores de la enzima de conversión de la angiotensina como los antagonistas de los receptores de la angiotensina II bloquean esta vía y reducen las resistencias totales periféricas y el trabajo miocárdico en último término1.
Péptidos natriuréticos
Los PN, fundamentalmente el péptido natriurético auricular (ANP), el péptido natriurético cerebral (BNP) y el péptido natriurético tipo C (CNP), favorecen la pérdida de sal y agua corporal por el organismo y suponen un mecanismo contrarregulador del sistema nervioso simpático y del SRAA. Tanto el ANP como el BNP, que se localizan en la aurícula y el ventrículo respectivamente, se liberan como respuesta al estiramiento de las fibras miocárdicas por la expansión de volumen, que incrementa la tensión de las paredes cardiacas y promueve la vasodilatación, la natriuresis y la diuresis. En cambio, el CNP, que se localiza principalmente en el sistema nervioso central y las células endoteliales, inhibe la secreción de renina, aldosterona y vasopresina. En la IC se produce un aumento de los PN para equilibrar los efectos de las hormonas vasoconstrictoras. Sin embargo, conforme la IC progresa, el efecto de los PN disminuye tanto por la hipoperfusión renal como por reducción de sus concentraciones3,4,15-18.
Los PN son degradados por la neprilisina, una metaloendopepti- dasa. Sin embargo, la neprilisina no solo degrada los PN (ANP, BNP, CNP), sino también otros péptidos como la endotelina, la sustancia P, la bradicinina y la angiotensina II a fragmentos inactivos, y en consecuencia reduce sus concentraciones. El sacubitrilo inhibe la neprili- sina y permite que los PN (ANP, CNP y en menor medida BNP) interactúen con sus receptores NPR-A y NPR-B y aumenten la cantidad intracitoplásmica de GMPc, lo que induce vasodilatación, disminución de la fibrosis/hipertrofia cardiaca y aumento de la natriuresis/diuresis19. Por lo tanto, la inhibición de la neprilisina aumenta las concentraciones de estos péptidos, en particular los PN, y promueve la vasodilatación, la natriuresis y la inhibición de la hipertrofia y la fibrosis, pero también de la angiotensina II, con lo que disminuyen los efectos beneficiosos de los PN3,4,15-18. En este contexto, la inhibición dual de la neprilisina con sacubitrilo junto con un inhibidor del SRAA vía el receptor de angiotensina II (valsartán) consigue el doble objetivo positivo de aumentar los PN y antagonizar el efecto de la angio- tensina II, lo que se ha traducido en una reducción de eventos, en comparación con la simple inhibición del SRAA20.
Inhibición del cotransportador de sodio-glucosa tipo 2Los inhibidores del cotransportador de sodio-glucosa tipo 2 (SGLT2) inicialmente se desarrollaron como fármacos hipogluce- miantes por su efecto glucosúrico al actuar sobre el túbulo contorneado proximal. Los SGLT2 también se encuentran en otros tejidos, aunque en menor concentración, y en diferentes ensayos clínicos se ha demostrado que inhibirlos reduce los ingresos por IC de la población con diabetes mellitus tipo 2, con o sin antecedentes de IC21, al igual que reducen el riesgo de hospitalización por IC o la mortalidad cardiovascular de los pacientes con IC, tengan diabetes o no22-24. En consecuencia, parece que los beneficios de los inhibidores del SGLT2 estarían mediados por mecanismos diferentes de su capacidad para reducir la glucemia y corregir los trastornos metabólicos asociados con la diabetes. De hecho, se han descrito múltiples mecanismos que explicarían su efecto protector contra la IC como, entre otros, la disminución de la glucotoxicidad, el empleo más eficiente de fuentes energéticas cardiacas, la disminución del volumen plasmático, el aumento de la diuresis, la reducción de la presión arterial, de la activación del sistema nervioso simpático, la rigidez arterial, la adiposidad, la inflamación, el estrés oxidativo y la fibrosis, la pérdida de peso, la protección renal, el aumento de la disponibilidad de óxido nítrico, la inhibición del intercambiador de sodio-hidrógeno (NHE) del sarco- lema, que disminuye la concentración intracelular de sodio en los cardiomiocitos, lo que tiene un efecto cardioprotector, así como la mejora del transporte de oxígeno, al aumentar la hemoglobina/hematocrito, independientemente de la disminución del volumen plasmático25-28.
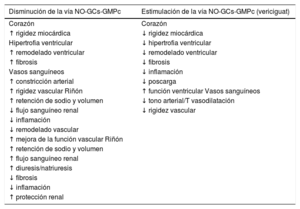
Vía óxido nítrico-guanilato ciclasa soluble-GMPcLa vía de señalización óxido nítrico-guanilato ciclasa soluble es clave en la regulación del sistema cardiovascular. La guanilato ciclasa soluble (GCs) sirve de receptor para el óxido nítrico derivado del endotelio y, tras su unión, la GCs cataliza la formación de la molécula de señalización GMPc, imprescindible para regular ciertas funciones celulares como el tono vascular, la proliferación, la fibrosis o la inflamación. En la IC se produce una disfunción de las células endoteliales que condiciona una disminución en la producción de óxido nítrico y una insuficiente estimulación de la GCs, lo que en último término se traduce en una reducción de la formación del GMPc. Esta reducción del GMPc provoca alteraciones en múltiples órganos. En el corazón, el déficit de GMPc se asocia con un aumento de la hipertrofia ventricular, un mayor remodelado, fibrosis y rigidez miocárdica; en el lecho vascular, con más vasoconstricción y rigidez de los vasos, y en el riñón, con una disminución del flujo sanguíneo renal y una mayor retención de sodio y agua. Por lo tanto, la reducción de GMPc contribuye tanto a la génesis como a la progresión de la IC (tabla 1 )29-40.
Se han investigado diferentes fármacos con el objetivo de modular la vía óxido nítrico-guanilato ciclasa soluble-GMPc:
- •
Activadores de la GCs dependientes del óxido nítrico. Aunque la infusión intravenosa de nitroglicerina está bien establecida como opción terapéutica para la IC aguda, no ha demostrado beneficio pronóstico tanto en fase aguda como crónica. Por otro lado, los inhibidores de la fosfodiesterasa 3 (milrinona) o 5 (sildenafilo) han mostrado mejoría hemodinámica, aunque con resultados dispares en pacientes con IC y fracción de eyección reducida.
- •
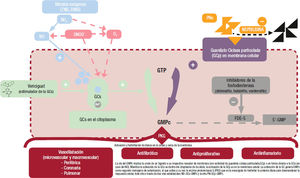
Vía PN-GCp. Se han evaluado diferentes PN sintéticos y análogos de los PN, y aunque en general conseguían una mejora en parámetros hemodinámicos y natriuresis, esto no se asociaba con una reducción en la morbimortalidad de los pacientes con IC y fracción de eyección reducida32. Sin embargo, como ya se ha comentado, la inhibición dual de la neprilisina y la angiotensina mostró un beneficio clínico en estos pacientes, con reducción del riesgo de muerte cardiovascular, muerte por cualquier causa y hospitalización por IC (figura 4)20.
- •
Moduladores de la GCs, activadores y estimuladores. Los activadores de GCs aumentan el GMPc mediante la activación directa e independiente del óxido nítrico de GCs y pueden ser particularmente efectivos en condiciones de aumento del estrés oxidativo y disfunción endotelial y, por tanto, reducción de óxido nítrico, pero esto se produce a expensas de un mayor riesgo de hipotensión. Por el contrario, los moduladores de la GCs (riociguat, vericiguat) mejoran la sensibilidad de la GCs al óxido nítrico endógeno y luego ejercen una acción más fisiológica, lo que posiblemente explique sus efectos neutrales en la presión arterial. El vericiguat es el primer fármaco que actúa por esta vía con probados beneficios clínicos en pacientes con IC (figura 4.)32.
 y el sacubitrilo (vía PN-GCp-GMPc). Figura elaborada con datos de McMurray20, Emdin32, Boerrigter35, Hulot37 y Sandner39,40 GCp: guanilato ciclasa particulada; GCs: guanilato ciclasa soluble; GMPc: guanosina monofosfato cíclico; NO: óxido nítrico; PN: péptidos natriuréticos.") Figura 4.
Figura 4.Efecto sinérgico entre el mecanismo de acción del vericiguat (vía NO-GCs-GMPc) y el sacubitrilo (vía PN-GCp-GMPc). Figura elaborada con datos de McMurray20, Emdin32, Boerrigter35, Hulot37 y Sandner39,40 GCp: guanilato ciclasa particulada; GCs: guanilato ciclasa soluble; GMPc: guanosina monofosfato cíclico; NO: óxido nítrico; PN: péptidos natriuréticos.
(0.23MB).
 y el sacubitrilo (vía PN-GCp-GMPc). Figura elaborada con datos de McMurray20, Emdin32, Boerrigter35, Hulot37 y Sandner39,40 GCp: guanilato ciclasa particulada; GCs: guanilato ciclasa soluble; GMPc: guanosina monofosfato cíclico; NO: óxido nítrico; PN: péptidos natriuréticos.")
Vericiguat es un estimulador oral de la GCs. Se une selectiva y específicamente a esta, lo que conduce a un aumento de la producción de GMPc dependiente de la concentración, esto es, que cuanto mayor es la concentración de vericiguat, mayor es el efecto en la producción de GMPc. En consecuencia, el vericiguat restablece la deficiencia relativa de GMPc en la vía de señalización óxido nítrico-guani- lato ciclasa soluble mediante la estimulación directa de la guanilato ciclasa soluble, de manera independiente y sinérgica con el óxido nítrico (figura 4). Esto se traduce en efectos positivos en diferentes órganos y sistemas del organismo. En el corazón, el aumento del GMPc mediante el tratamiento con vericiguat se asocia con una disminución de la rigidez miocárdica, la hipertrofia ventricular, el remodelado ventricular, la fibrosis, la respuesta inflamatoria y la poscarga. Todo ello finalmente mejora la función contráctil del corazón. En los vasos sanguíneos, el incremento de GMPc intracelular produce una reducción del tono arterial junto con vasodilatación y disminución de la rigidez vascular, la inflamación y el remodelado vascular, lo que en último término lleva a una mejoría de la función vascular. El vericiguat también aporta efectos beneficiosos renales mediante el aumento en las concentraciones intracelulares de GMPc, ya que disminuye la retención de sodio y agua, la fibrosis y la inflamación, a la vez que mejora el flujo sanguíneo renal y la diuresis/natriuresis, esto es, proporciona una mayor protección renal (tabla 1)36,41. El impacto de todos estos efectos beneficiosos se han visto en los resultados del ensayo clínico VICTORIA, en el que en más de 5.000 pacientes con IC y fracción de eyección reducida sintomática la adición de vericiguat al tratamiento médico estándar se tradujo en reducciones significativas de la variable combinada de muerte de causa cardiovascular o primera hospitalización por IC42.
| Disminución de la vía NO-GCs-GMPc | Estimulación de la vía NO-GCs-GMPc (vericiguat) |
|---|---|
| Corazón | Corazón |
| ↑ rigidez miocárdica | ↓ rigidez miocárdica |
| Hipertrofia ventricular | ↓ hipertrofia ventricular |
| ↑ remodelado ventricular | ↓ remodelado ventricular |
| ↑ fibrosis | ↓ fibrosis |
| Vasos sanguíneos | ↓ inflamación |
| ↑ constricción arterial | ↓ poscarga |
| ↑ rigidez vascular Riñón | ↑ función ventricular Vasos sanguíneos |
| ↑ retención de sodio y volumen | ↓ tono arterial/T vasodilatación |
| ↓ flujo sanguíneo renal | ↓ rigidez vascular |
| ↓ inflamación | |
| ↓ remodelado vascular | |
| ↑ mejora de la función vascular Riñón | |
| ↑ retención de sodio y volumen | |
| ↑ flujo sanguíneo renal | |
| ↑ diuresis/natriuresis | |
| ↓ fibrosis | |
| ↓ inflamación | |
| ↑ protección renal |
GCs: guanilato ciclasa soluble; GMPc: guanosina monofosfato cíclico; NO: óxido nítrico.
Aunque con ciertos matices en el orden de los fármacos, las diferentes guías de práctica clínica de 2021 para el tratamiento del paciente con IC (la europea, la actualización del American College of Cardiology y la canadiense) establecen que solo mediante un tratamiento integral con los fármacos que actúan por las diferentes vías (sistemas neurohormonales) de la etiopatogenia de la IC, pero que son complementarios entre sí, es posible mejorar el pronóstico de los pacientes con IC y fracción de eyección reducida1,43,44. Entre los distintos grupos farmacológicos recomendados, se encuentran los inhibidores del SRAA (inhibidores de la enzima de conversión de la angio- tensina, antagonistas del receptor de la angiotensina II y antagonistas de la aldosterona), los inhibidores del sistema nervioso simpático (BB), los inhibidores del SGLT2, los inhibidores de la neprilisina (sacu- bitrilo) y más recientemente los estimuladores de la guanilato ciclasa soluble (vericiguat)1,43,44.
ConclusionesLa IC es un síndrome complejo en el que, aunque inicialmente se ponen en marcha una serie de mecanismos compensatorios, estos resultan perjudiciales a largo plazo. Entre los mecanismos más importantes se encuentran los diferentes sistemas neurohormonales que — algunos por activación de vías deletéreas como el sistema nervioso simpático o el SRAA y otros por inhibición de vías protectoras (sistema de la neprilisina o la reducción del GMPc por inhibición de la vía óxido nítrico-GCs)— acaban produciendo un deterioro progresivo en diferentes órganos, principalmente corazón, lecho vascular y riñones. En consecuencia, solo mediante un tratamiento integral que tenga como objetivo mejorar el efecto negativo de todos estos sistemas neu-rohormonales, bien por exceso, bien por defecto, se podrá conseguir el mayor beneficio posible en el paciente con IC y fracción de eyección reducida.
Contribución de los autoresTodos los autores han contribuido significativamente al trabajo presentado en este artículo, contribuyendo en la concepción, diseño o adquisición de información, o en el análisis e interpretación de datos. Todos los autores han participado en la redacción o revisión del manuscrito y aceptan su publicación.
Conflictos de interesesA. García-Quintana ha recibido financiación por servicios de con- sultoría y conferencias de Bayer, Daiichi-Sankyo, Pfizer, AstraZeneca, Boehringer Ingelheim, Novartis y Rovi. A. Recio-Mayoral ha recibido financiación por servicios de consultoría y conferencias de Bayer, Janssen, MSD, Novartis y Vifor. J.M. Cepeda-Rodrigo no declara ningún conflicto de intereses en relación con este artículo. J.L. Zamorano ha recibido honorarios por ponencias de Bayer y Daiichi-Sankyo. J.R. González-Juanatey ha recibido compensaciones por asesoría y ponencias de Amgen, AstraZeneca, Bayer, Boehringer-Ingelheim, MSD, Daichii-Sankyo, Ferrer International, Novartis, Lilly, Sanofi y Servier.
AgradecimientosContent Ed Net (Madrid) proporcionó asistencia editorial en la redacción de este manuscrito con financiación de Bayer Hispania.