La miocardiopatía dilatada es familiar hasta en el 50% de los casos. Se han identificado más de 90 genes implicados en la enfermedad. Es una de las causas principales de trasplante cardiaco y se asocia con un riesgo aumentado de muerte súbita. La estratificación del riesgo de estos pacientes sigue siendo un reto. La identificación de la causa específica de la enfermedad es muy útil en la detección precoz de los familiares portadores. En muchos casos, el estudio genético aporta información pronóstica y puede condicionar la actitud terapéutica. La variabilidad fenotípica es amplia y depende del gen mutado, pero también del tipo de mutación identificada y otros factores genéticos y ambientales.
Palabras clave
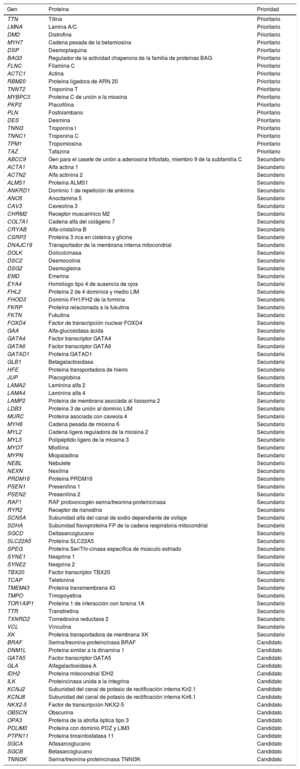
La miocardiopatía dilatada (MCD) se define como la dilatación y disfunción sistólica del ventrículo izquierdo o biventricular en ausencia de condiciones de carga anormal o enfermedad coronaria1. Es una de las principales causas de trasplante cardiaco y se asocia con un riesgo incrementado de muerte súbita. La prevalencia de la enfermedad se ha estimado en 1:2.500 individuos, aunque estudios recientes apuntan que podría ser mayor2. Se considera que la enfermedad es familiar hasta en el 50% de los casos, y en los últimos años se han identificado más de 90 genes implicados (tabla 1). La herencia en la mayoría de los casos es autosómica dominante, y son menos frecuentes la ligada al cromosoma X, la autosómica recesiva y la mitocondrial. Los genes implicados están relacionados con proteínas del sarcómero, el citoesqueleto, las uniones intercelulares, los canales iónicos y las proteínas mitocondriales.
Principales genes asociados con miocardiopatía dilatada. Se incluyen genes prioritarios, claramente asociados con la enfermedad y recomendados en las guías de práctica clínica. También se incluyen genes secundarios, relacionados esporádicamente y candidatos que surgen de la revisión sistemática de la literatura médica
| Gen | Proteína | Prioridad |
|---|---|---|
| TTN | Titina | Prioritario |
| LMNA | Lamina A/C | Prioritario |
| DMD | Distrofina | Prioritario |
| MYH7 | Cadena pesada de la betamiosina | Prioritario |
| DSP | Desmoplaquina | Prioritario |
| BAG3 | Regulador de la actividad chaperona de la familia de proteínas BAG | Prioritario |
| FLNC | Filamina C | Prioritario |
| ACTC1 | Actina | Prioritario |
| RBM20 | Proteína ligadora de ARN 20 | Prioritario |
| TNNT2 | Troponina T | Prioritario |
| MYBPC3 | Proteína C de unión a la miosina | Prioritario |
| PKP2 | Placofilina | Prioritario |
| PLN | Fosfolambano | Prioritario |
| DES | Desmina | Prioritario |
| TNNI3 | Troponina I | Prioritario |
| TNNC1 | Troponina C | Prioritario |
| TPM1 | Tropomiosina | Prioritario |
| TAZ | Tafazina | Prioritario |
| ABCC9 | Gen para el casete de unión a adenosina trifosfato, miembro 9 de la subfamilia C | Secundario |
| ACTA1 | Alfa actina 1 | Secundario |
| ACTN2 | Alfa actinina 2 | Secundario |
| ALMS1 | Proteína ALMS1 | Secundario |
| ANKRD1 | Dominio 1 de repetición de ankirina | Secundario |
| ANO5 | Anoctamina 5 | Secundario |
| CAV3 | Caveolina 3 | Secundario |
| CHRM2 | Receptor muscarínico M2 | Secundario |
| COL7A1 | Cadena alfa del colágeno 7 | Secundario |
| CRYAB | Alfa-cristalina B | Secundario |
| CSRP3 | Proteína 3 rica en cisteína y glicina | Secundario |
| DNAJC19 | Transportador de la membrana interna mitocondrial | Secundario |
| DOLK | Dolicolcinasa | Secundario |
| DSC2 | Desmocolina | Secundario |
| DSG2 | Desmogleína | Secundario |
| EMD | Emerina | Secundario |
| EYA4 | Homólogo tipo 4 de ausencia de ojos | Secundario |
| FHL2 | Proteína 2 de 4 dominios y medio LIM | Secundario |
| FHOD3 | Dominio FH1/FH2 de la formina | Secundario |
| FKRP | Proteína relacionada a la fukutina | Secundario |
| FKTN | Fukutina | Secundario |
| FOXD4 | Factor de transcripción nuclear FOXD4 | Secundario |
| GAA | Alfa-glucosidasa ácida | Secundario |
| GATA4 | Factor transcriptor GATA4 | Secundario |
| GATA6 | Factor transcriptor GATA6 | Secundario |
| GATAD1 | Proteína GATAD1 | Secundario |
| GLB1 | Betagalactosidasa | Secundario |
| HFE | Proteína transportadora de hierro | Secundario |
| JUP | Placoglobina | Secundario |
| LAMA2 | Laminina alfa 2 | Secundario |
| LAMA4 | Laminina alfa 4 | Secundario |
| LAMP2 | Proteína de membrana asociada al lisosoma 2 | Secundario |
| LDB3 | Proteína 3 de unión al dominio LIM | Secundario |
| MURC | Proteína asociada con caveola 4 | Secundario |
| MYH6 | Cadena pesada de miosina 6 | Secundario |
| MYL2 | Cadena ligera reguladora de la miosina 2 | Secundario |
| MYL3 | Polipéptido ligero de la miosina 3 | Secundario |
| MYOT | Miotilina | Secundario |
| MYPN | Miopaladina | Secundario |
| NEBL | Nebulete | Secundario |
| NEXN | Nexilina | Secundario |
| PRDM16 | Proteína PRDM16 | Secundario |
| PSEN1 | Presenilina 1 | Secundario |
| PSEN2 | Presenilina 2 | Secundario |
| RAF1 | RAF protooncogén serina/treonina-proteincinasa | Secundario |
| RYR2 | Receptor de rianodina | Secundario |
| SCN5A | Subunidad alfa del canal de sodio dependiente de voltaje | Secundario |
| SDHA | Subunidad flavoproteína FP de la cadena respiratoria mitocondrial | Secundario |
| SGCD | Deltasarcoglucano | Secundario |
| SLC22A5 | Proteína SLC22A5 | Secundario |
| SPEG | Proteína Ser/Thr-cinasa específica de músculo estriado | Secundario |
| SYNE1 | Nesprina 1 | Secundario |
| SYNE2 | Nesprina 2 | Secundario |
| TBX20 | Factor transcriptor TBX20 | Secundario |
| TCAP | Teletonina | Secundario |
| TMEM43 | Proteína transmembrana 43 | Secundario |
| TMPO | Timopoyetina | Secundario |
| TOR1AIP1 | Proteína 1 de interacción con torsina 1A | Secundario |
| TTR | Transtiretina | Secundario |
| TXNRD2 | Tiorredoxina reductasa 2 | Secundario |
| VCL | Vinculina | Secundario |
| XK | Proteína transportadora de membrana XK | Secundario |
| BRAF | Serina/treonina-proteincinasa BRAF | Candidato |
| DNM1L | Proteína similar a la dinamina 1 | Candidato |
| GATA5 | Factor transcriptor GATA5 | Candidato |
| GLA | Alfagalactosidasa A | Candidato |
| IDH2 | Proteína mitocondrial IDH2 | Candidato |
| ILK | Proteincinasa unida a la integrina | Candidato |
| KCNJ2 | Subunidad del canal de potasio de rectificación interna Kir2.1 | Candidato |
| KCNJ8 | Subunidad del canal de potasio de rectificación interna Kir6.1 | Candidato |
| NKX2-5 | Factor de transcripción NKX2-5 | Candidato |
| OBSCN | Obscurina | Candidato |
| OPA3 | Proteína de la atrofia óptica tipo 3 | Candidato |
| PDLIM3 | Proteína con dominio PDZ y LIM3 | Candidato |
| PTPN11 | Proteína tirosinfosfatasa 11 | Candidato |
| SGCA | Alfasarcoglucano | Candidato |
| SGCB | Betasarcoglucano | Candidato |
| TNNI3K | Serina/treonina-proteincinasa TNNI3K | Candidato |
La estratificación del riesgo de los pacientes con MCD sigue siendo un reto. Aunque diversos factores se han asociado con un incremento en el riesgo de progresión de enfermedad y muerte súbita, su utilidad clínica es escasa y el pronóstico se suele determinar por el deterioro de la clase funcional y la afección grave según técnicas de imagen3. La importancia de determinar el genotipo se ha establecido en algunos casos específicos, como las mutaciones en LMNA que se han asociado con peor pronóstico de la enfermedad4,5. Dada la gran variedad de causas de MCD, es probable que la generalización del riesgo de mal pronóstico en estos pacientes no sea adecuada y la identificación de la etiología específica sea necesaria para una adecuada estratificación pronóstica y un correcto abordaje terapéutico. El conocimiento de la causa de la enfermedad permite además el diagnóstico temprano de los familiares portadores que pueden requerir un seguimiento estrecho y un tratamiento más precoz, así como evitar el seguimiento innecesario de familiares no afectados6.
Esta revisión pretende describir la utilidad del estudio genético en la valoración pronóstica de los pacientes con MCD. Como se verá a continuación, pueden observarse diferencias pronósticas entre diferentes genes, y entre distintas mutaciones que afectan a un mismo gen. De hecho, en cualquiera de los genes evaluados puede haber variantes que ni siquiera producen enfermedad. Incluso dentro de una familia la misma mutación identificada puede dar lugar a fenotipos diferentes por la influencia de otros factores genéticos y ambientales, el sexo, la edad, etc. Por eso es esencial una evaluación individualizada de cada variante genética que se identifique y realizar un estudio sistemático genético y clínico de las familias. De algunos de los genes asociados, la información es escasa, ya que con frecuencia en las publicaciones los datos clínicos de los pacientes y las familias aportados son muy pocos y es difícil extraer conclusiones sobre la posible relevancia de las mutaciones identificadas.
MCD POR MUTACIONES EN GENES SARCOMÉRICOSTitinaEl gen TTN codifica la proteína de mayor tamaño expresada en el corazón y es el implicado con más frecuencia en la MCD. Esta proteína se extiende desde el disco Z hasta la línea M en el centro del sarcómero y se encarga de mantener su integridad estructural. Las mutaciones radicales en TTN (mutaciones que generan truncamientos, como cambios de pauta de lectura, mutaciones sin sentido o que afectan al proceso de corte y empalme del ARN) se han asociado con MCD con un patrón de herencia autosómico dominante y explican un 14-25% de los casos de esta enfermedad7,8. En general, las mutaciones patogénicas descritas se localizan en la banda A de la proteína y afectan a exones ampliamente expresados en las distintas isoformas9. Otros fenotipos asociados son la distrofia muscular tibial y algunas formas recesivas, como la distrofia muscular de cinturas de tipo 2 y la miopatía de aparición temprana con miocardiopatía asociada. Los estudios iniciales de pacientes con MCD y truncamientos en TTN no encontraron diferencias pronósticas entre los pacientes portadores de este tipo de variantes y los no portadores, aunque al comparar por sexos, los varones sufrieron eventos a una edad menor7. Posteriormente, otros autores demostraron una mayor prevalencia de arritmias ventriculares en los portadores (el 64 frente al 21%), aunque con un tamaño muestral pequeño10. Estudios más amplios han documentado un riesgo de fibrilación auricular y/o taquicardia ventricular 3 veces mayor tras un ajuste por los factores de riesgo convencionales. Se evidenciaron arritmias en el 46% de los pacientes con truncamientos en TTN, frente al 33% de los pacientes no portadores11. Otros estudios han mostrado un pronóstico más benigno, comparado con MCD por variantes en LMNA o estudio genético negativo. La enfermedad fue menos grave en su presentación y tuvo una evolución más favorable, con mejor respuesta al tratamiento médico12. En un reciente trabajo presentado por nuestro grupo en el congreso de la Sociedad Europea de Cardiología, que incluía para el análisis la información de más de 500 portadores y familiares afectados con truncamientos en TTN, se observó una elevada incidencia de muerte cardiovascular a partir de los 30 años de edad. Es importante destacar que la incidencia fue mayor en varones que en mujeres y que la mitad de los eventos se debieron a muerte súbita13.
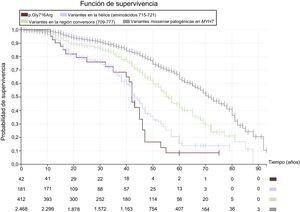
Proteínas del sarcómeroLos genes que codifican las proteínas del sarcómero se han asociado fundamentalmente con el desarrollo de miocardiopatía hipertrófica (MCH) con patrón de herencia autosómico dominante14, aunque la mayoría también están implicados en la MCD. Los genes más frecuentemente afectados son MYH7, TNNT2 y TPM1, mientras que las variantes en MYBPC3 son más raras. La presencia de estas mutaciones se ha relacionado con una presentación precoz de MCD, con aparición de insuficiencia cardiaca a edad temprana sin evidencia previa de hipertrofia ni desorganización miofibrilar, lo que indica enfermedad primaria15. Hay trabajos que han mostrado una elevada tasa de eventos (muerte y trasplante) a partir de los 50 años, independientemente de la fracción de eyección, aunque no encontraron diferencias en la supervivencia a largo plazo16. Para las variantes en MYH7, se han evidenciado diferencias pronósticas en función de su localización en la proteína. De esta forma, las variantes missense que se localizan en la región conversora (aminoácidos 709-777) se han relacionado con una presentación precoz de la enfermedad y elevada prevalencia de eventos17. Pero incluso dentro de esta región se observan diferencias importantes en evolución y pronóstico con mutaciones diferentes. En particular, las variantes descritas en una hélice alfa que abarca desde el aminoácido 715 al 722 se asocian con un pronóstico especialmente desfavorable, con alta incidencia de muerte súbita de jóvenes y aparición de insuficiencia cardiaca con muerte por esta causa o trasplante antes de los 50 años (figura 1). También la región de unión a la actina (aminoácidos 526-557) parece ser funcionalmente muy relevante, con 64 portadores identificados de 30 familias diferentes, la mayoría diagnosticados a edades tempranas y con disfunción ventricular moderada (datos no publicados). Aunque no es posible establecer un pronóstico general para las mutaciones en TTNT2, cuando producen MCD suelen asociarse con grandes penetrancia y frecuencia de eventos adversos18. En cuanto a las variantes en TPM1, en la región central flexible del extremo C-terminal (aminoácidos 81-258) se han descrito varias mutaciones missense relacionadas con MCD. En muchas de ellas se describe a portadores en edad pediátrica, algunos con eventos principalmente por insuficiencia cardiaca19. Es importante destacar que, aunque las mutaciones en MYBPC3 en general no conllevan mal pronóstico, pueden dar lugar a fenotipos graves si se presentan en homocigosis o heterocigosis compuesta.
. Se comparan los eventos en portadores de variantes missense patogénicas en MYH7 (gris), variantes en la región conversora 709-777 (verde), variantes en la hélice 715-721 (azul) y portadores de la variante p.Gly716Arg (granate). Se observan diferencias significativas en la mortalidad cardiovascular entre las regiones, con un pronóstico peor para las variantes localizadas en la hélice como p.Gly716Arg y una supervivencia muy baja a los 50 años. Con las variantes de peor pronóstico, el porcentaje de pacientes con diagnóstico inicial o evolución a miocardiopatía dilatada es superior que la de las variantes con mejor pronóstico. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Curvas de Kaplan-Meier que analizan la supervivencia libre de muerte cardiovascular (muerte súbita, descarga apropiada del desfibrilador, muerte por insuficiencia cardiaca o trasplante y muerte de otra causa cardiovascular). Se comparan los eventos en portadores de variantes missense patogénicas en MYH7 (gris), variantes en la región conversora 709-777 (verde), variantes en la hélice 715-721 (azul) y portadores de la variante p.Gly716Arg (granate). Se observan diferencias significativas en la mortalidad cardiovascular entre las regiones, con un pronóstico peor para las variantes localizadas en la hélice como p.Gly716Arg y una supervivencia muy baja a los 50 años. Con las variantes de peor pronóstico, el porcentaje de pacientes con diagnóstico inicial o evolución a miocardiopatía dilatada es superior que la de las variantes con mejor pronóstico. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
La actina, que codifica el gen ACTC1, es el principal componente de los filamentos finos del sarcómero. Se han descrito pocas mutaciones en este gen, algunas de ellas vinculadas a un fenotipo solapado de MCH no compactada y MCD con patrón autosómico dominante. Es de destacar la presencia de defectos septales en relación con algunas de las variantes descritas. La evolución a insuficiencia cardiaca se relacionó con la presencia de disfunción diastólica y fenotipo restrictivo20.
MCD POR MUTACIONES EN PROTEÍNAS DEL CITOESQUELETOLaminaEl gen LMNA codifica 2 proteínas, laminas A y C, componentes de la cara interna de la membrana nuclear. Las mutaciones en este gen se han asociado con un grupo de enfermedades con patrón autosómico dominante que en conjunto se denominan laminopatías e incluyen MCD, distrofia muscular de Emery Dreifuss, lipodistrofia familiar parcial, distrofia muscular de Limb Girdle, enfermedad de Charcot-Marie-Tooth y progeria. La afección cardiaca se caracteriza por alto riesgo de muerte súbita, y son frecuentes los trastornos de conducción y las arritmias ventriculares, que suelen preceder a la aparición de disfunción ventricular21. Las arritmias supraventriculares (fibrilación auricular) también son frecuentes. La penetrancia es muy alta (casi el 100% de los portadores de mutaciones patogénicas tendrán la enfermedad a los 60 años). Se recomienda considerar un bajo umbral para el implante de desfibrilador, sobre todo para los pacientes que requieran implante de marcapasos22,23. Aunque las mutaciones más frecuentemente descritas son de tipo missense y se localizan en el dominio central de la proteína, también se han descrito variantes de tipo truncamiento. El pronóstico es peor en los portadores de mutaciones patogénicas respecto a los no portadores, aunque hay que tener en cuenta que no todas las mutaciones son iguales y algunas de ellas son variantes raras que no producen enfermedad. Se han definido 4 factores que aumentan de manera independiente el riesgo de arritmias de los portadores: la presencia de taquicardia ventricular no sostenida, la fracción de eyección < 45%, el sexo masculino y las mutaciones que no son missense (nonsense, frameshift y splicing)22. Sin embargo, nuestro grupo ha evaluado la información procedente de más de 1.000 portadores y familiares con mutaciones en LMNA sin observar diferencias relevantes entre varones y mujeres en cuanto a la presentación de eventos cardiovasculares (datos no publicados). Por otro lado, los resultados preliminares de un estudio multicéntrico que se está realizando en nuestro medio no aportan diferencias pronósticas entre mutaciones que causan truncamiento y missense24. Estos datos indican que se debería revisar los criterios utilizados actualmente para indicar el implante de desfibrilador a estos pacientes, especialmente considerando que la muerte súbita de pacientes con fracción de eyección > 45% no es infrecuente25.
DesminaEl gen DES codifica la proteína citoplásmica desmina, que es el principal componente de los filamentos intermedios. Las mutaciones en este gen se han asociado con MCD, trastornos de la conducción y debilidad muscular progresiva y elevada penetrancia. Existen formas tanto autosómicas recesivas como dominantes y la mayoría de las variantes patogénicas identificadas son de cambio de sentido. La prevalencia de la enfermedad es 1:10.000 aproximadamente26. En el corazón, los trastornos de la conducción suelen aparecer antes que las alteraciones del miocardio y es frecuente un fenotipo restrictivo27. Se ha descrito que hasta el 50% de los portadores presentan miocardiopatía y alrededor del 60% tienen trastornos de la conducción y arritmias ventriculares. La ausencia de miopatía no descarta la enfermedad. Si bien el bloqueo auriculoventricular es característico, se ha descrito muerte arrítmica en varios casos, algunos de ellos portadores de marcapasos28.
DistrofinaLa distrofina es una proteína citoesquelética de gran tamaño que se encuentra en la superficie interior de las células musculares y se codifica por el gen DMD. Existen 3 fenotipos asociados con el gen: las distrofias musculares de Duchenne (DMD) y Becker (BMD), y la MCD ligada a cromosoma X. La diferencia entre los fenotipos se relaciona con la gravedad de la afección muscular. En cuanto a la MCD, esta puede presentarse tanto en varones como en mujeres portadores de variantes patogénicas, con una media de edad al diagnóstico entre los 20 y los 40 años en los varones y un poco más tarde en las mujeres. La progresión de la enfermedad a estadios graves suele ser rápida en los varones, mientras que en las mujeres puede llevar varios años. El deterioro cardiaco se presenta en un 60-75% de los casos y está en relación con degeneración difusa y fibrosis de los ventrículos, especialmente en las regiones inferolaterales y en el tejido de conducción. La presencia de arritmias auriculares y ventriculares es frecuente. Respecto al tipo de mutación asociada, más de 2 de cada 3 casos son portadores de mutaciones tipo deleción de 1 o más exones. Se localizan principalmente en una región determinada entre los exones 45 y 53. Se han descrito duplicaciones parciales del gen en una pequeña proporción de individuos afectados (5-15%). Las mutaciones de tipo missense se han descrito en casos infrecuentes y por lo general afectan a regiones cardioespecíficas de la proteína. De hecho, más de la mitad de las mutaciones de este tipo relacionadas con MCD se localizan en el dominio de unión a la actina de DMD29,30.
EmerinaEl gen EMD se localiza en el cromosoma X y codifica la emerina, una proteína de la membrana nuclear rica en serina de la familia de proteínas nucleares asociadas con la lamina. Las mutaciones en este gen se asocian con la distrofia muscular de Emery-Dreifuss ligada a X, que se caracteriza por contracturas articulares de comienzo temprano (infancia o adolescencia), debilidad y atrofia progresiva de las extremidades y afección cardiaca con trastornos de la conducción, arritmias ventriculares y MCD. Aunque la muerte súbita puede ser secundaria a asistolia y por ello se puede prevenir con el implante de marcapasos, en algunos casos puede estar en relación con el desarrollo de fibrosis por la miocardiopatía y sería necesario implantar un desfibrilador31,32. Las mujeres portadoras suelen presentar un fenotipo más leve o no contraen enfermedad. La mayoría de las mutaciones patogénicas son de tipo radical, lo que resulta en una ausencia total de síntesis de emerina normal en el núcleo, pero también producen MCD mutaciones que producen el cambio de un único aminoácido. Un ejemplo interesante es la variante Val26Ala. En un estudio realizado en España que evaluó la presencia de alteraciones genéticas en pacientes trasplantados por MCD, se identificó esta variante en múltiples casos índice que se habían sometido a trasplante en 2 hospitales de Madrid, todos ellos originarios de una población de Canarias en la que desde hace tiempo se detectan múltiples casos de la enfermedad. En estos pacientes, no hay datos evidentes de miopatía o trastornos de la conducción, que son frecuentes en otras mutaciones de este gen. Al parecer, las mujeres portadoras heterocigotas no muestran signos de cardiopatía33.
MCD POR MUTACIONES EN GENES DESMOSÓMICOSLos desmosomas son proteínas que mantienen la integridad estructural de los contactos entre las células adyacentes mediante el anclaje a la placa desmosómica. Las mutaciones en estos genes se relacionan principalmente con el desarrollo de miocardiopatía arritmogénica (MCA), aunque puede haber formas de predominio izquierdo o biventriculares que son indistinguibles de la MCD. Los genes más frecuentemente relacionados con este fenotipo son DSP y PKP2, aunque otros genes desmosómicos también pueden asociarse34. La mayoría de los casos tienen un patrón de herencia autosómico dominante, pero puede haber algunas formas recesivas (síndrome de Carvajal). Los casos descritos tenían mayor incidencia de arritmias ventriculares y un mayor riesgo de muerte súbita, independientemente de la fracción de eyección35. La MCA de predominio izquierdo es una entidad a menudo infradiagnosticada, por el solapamiento con otras miocardiopatías y entidades como las miocarditis o la taquicardia ventricular idiopática. Debe sospecharse en presencia de arritmias de origen izquierdo y ondas T invertidas a nivel inferolateral36.
OTROS GENES MENOS PREVALENTES RELACIONADOS CON LA MCDGen RBM20El gen RBM20 codifica un miembro de la familia de proteínas SR (proteínas ricas en serina/arginina) que regula el splicing alternativo de diferentes genes, de los que el de la titina el más destacable. Las mutaciones identificadas en este gen hasta el momento se relacionan con MCD con patrón autosómico dominante. La mayoría de ellas son variantes missense que se localizan en 2 regiones funcionalmente muy relevantes, la región rica en Arg-Ser (exón 9) y el dominio en dedos de cinc (exón 14), aunque se han identificado otras variantes distribuidas por todo el gen. Algunos de los pacientes incluidos mostraron un pronóstico adverso con altas incidencias de muerte súbita, insuficiencia cardiaca y trasplante37,38. Otros trabajos no han encontrado diferencias en la supervivencia o la incidencia de arritmias ventriculares en los portadores, aunque en poblaciones con una baja tasa de eventos39. Se ha señalado que la localización de la variante en el gen podría determinar diferentes subtipos de enfermedad, aunque de momento esta hipótesis requiere confirmación.
Gen FLNCEl gen FLNC codifica una proteína citoplásmica de tipo unión a la actina. Las primeras mutaciones descritas se asociaron con miopatía miofibrilar esquelética, pero datos recientes indican que las miocardiopatías podrían ser el principal fenotipo clínico relacionado, con un patrón de herencia autosómico dominante. Un estudio multicéntrico liderado por nuestro grupo ha descubierto recientemente que las variantes de tipo truncamiento en este gen se asocian con un fenotipo solapado de MCD y MCA del ventrículo izquierdo, con alta penetrancia. Es característica la afección del ventrículo izquierdo casi exclusivamente, con dilatación y disfunción sistólicas que pueden ser ligeras, zonas extensas de fibrosis intramiocárdica en la pared del ventrículo izquierdo, alta frecuencia de arritmias ventriculares, ausencia de miopatía esquelética y creatincinasa normal. La incidencia de muerte súbita fue alta, principalmente a partir de los 40 años de edad, incluso en pacientes con disfunción ventricular no grave40. Recientemente, también se han relacionado variantes missense de este gen con miocardiopatía restrictiva y MCH, aunque el nivel de la evidencia es menor.
Gen BAG3El regulador de la actividad chaperona de la familia de proteínas BAG está codificado por el BAG3. Esta proteína presenta actividad antiapoptótica y tiene gran expresión en el músculo esquelético y cardiaco, donde se localiza en el disco Z. Son pocas las mutaciones de este gen descritas, dado que su asociación con fenotipos cardiovasculares en humanos es reciente41. Tanto deleciones de exones como mutaciones radicales (y con menos frecuencia algunas de cambio de sentido) se han asociado con la aparición de MCD con herencia autosómica dominante, y algunas de ellas producen un fenotipo grave42. Algunas mutaciones de cambio de sentido se han relacionado con miopatía miofibrilar, también con herencia autosómica dominante.
Gen PLNEl gen PLN codifica la proteína fosfolambano, que se encuentra en la membrana del retículo sarcoplásmico. La asociación de este gen con la MCD proviene de la mutación p.Arg14del, que tiene efecto fundador en los Países Bajos. Esta variante se ha relacionado con el desarrollo de MCA de predominio izquierdo y formas arrítmicas de MCD con patrón autosómico dominante, con una alta tasa de arritmias ventriculares malignas comparable a la de las mutaciones en LMNA y aparición de insuficiencia cardiaca avanzada a edad temprana43. La escasa amplitud de la onda R se ha definido como un marcador precoz de afección en portadores, que se ha correlacionado con la presencia de realce tardío en la resonancia magnética. Otros trabajos señalan una baja penetrancia de la enfermedad y fenotipos más leves, especialmente cuando las variantes son de tipo truncamiento44. También se han descrito algunas variantes asociadas con el desarrollo de MCH.
Gen SCN5AEl gen SCN5A codifica el canal de sodio dependiente del voltaje resistente a tetradotoxina, conocido como Nav1.5. La mayoría de las mutaciones descritas se relacionan con los síndromes de QT largo y de Brugada con patrón de herencia autosómico dominante. Otros fenotipos relacionados con el gen son la enfermedad progresiva del sistema de conducción (enfermedad de Lev-Lenègre), enfermedad del nódulo sinusal y fibrilación auricular. Un pequeño porcentaje de mutaciones en este gen se han asociado con MCD; la mejor caracterizada es p.Arg222Gln, de la que se ha demostrado que cosegrega con el fenotipo en múltiples familias de distintos países45. La mayoría de las mutaciones vinculadas a MCD son de tipo missense y se localizan en los segmentos transmembrana S3 y S4, dominios muy conservados. No está claro el mecanismo fisiopatológico por el que estas variantes se asocian con miocardiopatía, aunque existen varias hipótesis46.
Gen LAMP2El gen LAMP2 codifica una proteína de la membrana lisosomal y es la causa de la enfermedad de Danon o glucogenosis tipo IIb. Se han descrito múltiples mutaciones asociadas al desarrollo de miocardiopatía que se expresa de forma muy diferente en ambos sexos. Los varones (hemicigotos) adquieren en la infancia MCH con hipertrofia muy grave y evolución precoz a disfunción sistólica, mientras que en las mujeres el diagnóstico inicial suele ser MCD. La presencia de preexcitación en el electrocardiograma y las alteraciones de la conducción son comunes, y también se ha descrito la asociación con miopatía, retinopatía y alteraciones cognitivas, fundamentalmente en los varones47. El pronóstico es más grave en los varones, que suelen fallecer antes de los 30-40 años, pero el cuadro es también muy grave en las mujeres, en las que constituye la forma de MCD de peor pronóstico.
Genes mitocondrialesLas enfermedades mitocondriales pueden ser secundarias a mutaciones en el ADN mitocondrial, con herencia matrilineal. Sin embargo, la mayor parte de los genes implicados en mantener la estructura y la función de las mitocondrias se encuentran en el ADN nuclear, por lo que muestran patrones de herencia mendelianos, habitualmente autosómica recesiva. La presencia de trastornos del sistema nervioso central y de enfermedad multisistémica orienta hacia el diagnóstico de enfermedad mitocondrial. Entre los síndromes asociados con MCD, destaca el síndrome de Barth producido por mutaciones en el gen TAZ, que codifica la proteína tafazzina, implicada en el metabolismo de la cardiolipina mitocondrial. Clínicamente se caracteriza por miocardiopatía (dilatada, hipertrófica y no compactada), fibroelastosis endocárdica, miopatía esquelética, retraso del crecimiento, neutropenia y aciduria orgánica. El patrón de herencia está ligado al X y las mujeres suele ser portadoras asintomáticas48.
Influencia de factores adicionales en la MCD familiarLa variabilidad fenotípica y la penetrancia incompleta observadas en los pacientes con MCD afectados por una misma alteración genética indican que frecuentemente hay otros factores que pueden modificar la expresión y el pronóstico de la enfermedad (de manera tanto favorable como desfavorable), entre otros, variantes genéticas adicionales, modificadores ambientales y factores epigenéticos.
Para la mayoría de los genes asociados con MCD (LMNA, RBM20 y genes sarcoméricos), se ha observado un predominio de pacientes varones49. Además, para algunos genes se han observado importantes diferencias en la evolución y el pronóstico en función del sexo50. Es fácil explicar estas diferencias con los genes que se encuentran en el cromosoma X (como DMD o EMD), pero las causas de estas diferencias para genes que se encuentran en otros cromosomas por el momento no se han explicado satisfactoriamente.
Aunque el ejercicio físico habitualmente es recomendable para los pacientes con insuficiencia cardiaca, puede condicionar un mayor riesgo arrítmico para los pacientes con etiologías específicas. En los portadores de mutaciones en genes desmosómicos, se ha demostrado que el entrenamiento de resistencia aumenta la penetrancia de enfermedad y el riesgo de insuficiencia cardiaca avanzada y arritmias51. En pacientes con MCH y mutaciones en genes sarcoméricos, el ejercicio intenso se ha relacionado con un diagnóstico más precoz, aunque no se han identificado diferencias pronósticas relevantes52. También en los portadores de LMNA se ha evidenciado un mayor riesgo de eventos en pacientes que habían realizado ejercicio de alta intensidad, incluso aunque se hubiera reducido varios años antes del diagnóstico53.
Varios trabajos han demostrado que la expresión de la MCD familiar también puede modularse por otros factores ambientales, entre ellos las miocarditis, los déficit nutricionales y los agentes citotóxicos54–56. El alcohol se considera un importante factor etiológico en esta enfermedad y trabajos recientes han demostrado una elevada predisposición genética a la enfermedad en pacientes con MCD alcohólica57.
CONCLUSIONES Y PERSPECTIVAS FUTURAS SOBRE EL PAPEL DE LA GENÉTICA EN EL DIAGNÓSTICO Y LA ESTRATIFICACIÓN DEL RIESGO DE LA MCDLos avances que se han producido en el conocimiento de las bases genéticas de la MCD nos han permitido demostrar que lo que hasta ahora habíamos denominado MCD «idiopática» tiene una causa genética identificable en un elevado porcentaje de pacientes. Un estudio genético completo permite encontrar 1 o varias variantes que explican la enfermedad en aproximadamente un 50% de los casos que no tienen otra causa identificable, y se llega a más de un 70% cuando la enfermedad tiene una presentación familiar. Se ha confirmado también que la MCD idiopática no es una única enfermedad, sino un conjunto de enfermedades que tienen diferentes etiologías, evolución y pronóstico.
Por una parte, estos hallazgos aumentan la complejidad del abordaje diagnóstico de los pacientes con esta enfermedad, pero al mismo tiempo constituyen un avance esencial para el desarrollo de abordajes individualizados o personalizados, tanto diagnósticos como de pronóstico y tratamiento.
El diagnóstico precoz de los familiares portadores permite su adecuado seguimiento y actuar para detener el avance de la enfermedad. En los pacientes afectados, la identificación de la causa específica de la MCD aporta en muchos casos información sobre el riesgo de progresión de la enfermedad y muerte súbita, lo que puede tener importantes implicaciones terapéuticas.
La interpretación de los resultados de los estudios genéticos es compleja e implica la colaboración de equipos expertos de biólogos moleculares, genetistas y especialistas (en este caso cardiólogos). No basta con identificar 1 o varias variantes genéticas asociadas con la enfermedad; sino que es necesario reunir toda la información disponible sobre dichas variantes para que se pueda llegar a conclusiones sobre la historia natural de las diferentes formas de enfermedad que producen. El esfuerzo requerido para alcanzar este objetivo es grande, ya que existen muchas variantes genéticas con consecuencias y gravedades diferentes en cada uno de los múltiples genes asociados con la enfermedad. Hasta la fecha, la mayor parte de los estudios han buscado la simplificación y han comparado casos «con y sin mutaciones» o casos con mutaciones en distintos genes, lo que no es suficiente.
El fenotipo y el riesgo dependen del gen, pero sobre todo de la mutación concreta identificada (según el tipo, la región afectada, etc.) y la presencia de otros factores genéticos y/o ambientales. Casi en todos los genes se puede encontrar variantes de mayor y menor riesgo y variantes que ni siquiera producen enfermedad; por lo que limitarse a hablar de genes de alto y bajo riesgo es un error. Hoy se puede empezar a analizar el efecto de variantes individuales, o al menos de grupos de variantes que comparten características y efectos funcionales similares. Esta información se debe integrar con el conocimiento clínico disponible mediante una revaluación de los criterios diagnósticos y pronósticos de la enfermedad. En las guías de práctica clínica vigentes hay ejemplos, ya comentados, sobre cómo el conocimiento de la variante genética causante de la enfermedad puede modificar radicalmente la aproximación clínica a los pacientes afectados. En los próximos años se verá que estos ejemplos se multiplican y la norma será establecer criterios diagnósticos, pronósticos y terapéuticos respaldados por el conocimiento previo de la naturaleza específica de la enfermedad en consideración, ya sea de causa monogénica, poligénica o multifactorial. Este progreso requerirá un mayor conocimiento sobre las bases genéticas y moleculares de la MCD y un estudio específico de la relación entre variantes genéticas, evolución, pronóstico y respuesta a diferentes estrategias de prevención y tratamiento en cada una de las formas específicas de la enfermedad.
CONFLICTO DE INTERESESL. Monserrat es accionista de Health in Code S.L.