La amiloidosis cardiaca (AC) se produce por depósito de fibras de amiloide en el miocardio. Las formas más frecuentes son la amiloidosis por cadenas ligeras (AL) y por transtiretina (ATTR). Nuestro objetivo es describir la experiencia en el diagnóstico, el tratamiento y el pronóstico en un centro especializado español.
MétodosSe incluyó a todos los pacientes diagnosticados de AC en el Hospital Puerta de Hierro Majadahonda desde mayo de 2008 a septiembre de 2018 y se analizaron sus características clínicas, su evolución y su supervivencia.
ResultadosSe incluyó a 180 pacientes con AC, de los que 64 (36%) tenían AL (el 50% varones; edad, 65±11 años) y 116, ATTR (el 72% varones; edad, 79±11 años; 18 con ATTR hereditaria). La forma de presentación más frecuente fue la insuficiencia cardiaca en ambos grupos (el 81% con AL y el 45% con ATTR; p <0,01). Otras formas de presentación en pacientes con ATTR fueron arritmias auriculares (16%), trastornos de conducción (6%) e incidental (6%). Ya tenían otro diagnóstico establecido 70 pacientes (40%). Se pudo diagnosticar de manera no invasiva al 75% de los pacientes con ATTR. A pesar de que el retraso diagnóstico fue superior en la ATTR (2,8±4,3 frente a 0,6±0,7 años; p <0,001), la mortalidad fue mayor en los pacientes con AL (el 48 frente al 32%; p=0,028). El tipo de AL (HR=6,16; IC95%, 1,56-24,30; p=0,01), el sexo femenino (HR=2,35; IC95%, 1,24-4,46; p=0,01) y la clase funcional de la NYHA III-IV (HR=2,07; IC95%, 1,11-3,89; p=0,02) fueron predictores independientes de la mortalidad.
ConclusionesLa AC constituye un reto en la práctica clínica, con gran variabilidad en su presentación en función del subtipo y con un retraso diagnóstico y una mortalidad elevados. Son necesarias mejoras en el diagnóstico temprano y el tratamiento de estos pacientes.
Palabras clave
Vídeo del artículo
El término amiloidosis cardiaca (AC) hace referencia a la afección del corazón como consecuencia del depósito de fibras amiloides1. En nuestro medio, los subtipos más frecuentes de AC son la amiloidosis por cadenas ligeras (AL), consecuencia del depósito de cadenas ligeras procedentes de una discrasia de células plasmáticas, y la debida al depósito de la proteína transtiretina (TTR) o amiloidosis por TTR (ATTR). De esta hay 2 formas: la variante hereditaria (ATTRh) y el subtipo no hereditario (ATTRwt) previamente denominada amiloidosis senil1,2.
Clásicamente, se consideraba la forma AL como el subtipo de amiloidosis más prevalente en los países desarrollados3. Sin embargo, la utilización de técnicas diagnósticas no invasivas4 y el envejecimiento progresivo de la población hacen que hoy se considere la ATTR como la forma más frecuente de AC5.
La AC es una entidad hasta ahora infradiagnosticada que presenta un importante retraso diagnóstico6. Parte de este retraso se debe a la consideración tradicional de la AC como una enfermedad no tratable, así como a la complejidad de su diagnóstico, incluida la necesidad de demostración histológica2,5. Sin embargo, actualmente existe la posibilidad de realizar el diagnóstico de AC sin necesidad de demostración histológica en un grupo importante de pacientes con AC, y se han producido avances muy importantes tanto en las técnicas de diagnóstico como en el tratamiento7-13.
En la última década hemos asistido al desarrollo de nuevos tratamientos con resultados prometedores tanto en ATTR como en la AL7-10. Algunos de estos fármacos han mostrado ya resultados muy satisfactorios en ensayos clínicos10,11. Por ello se hace imprescindible identificar a los individuos que podrán beneficiarse de los nuevos tratamientos, sobre todo en estadios precoces, que es cuando son más efectivos.
Hasta ahora, la experiencia en el diagnóstico y el tratamiento de los pacientes con AC era escasa y confinada a unos pocos centros. Con el aumento del número de pacientes diagnosticados y el desarrollo de nuevos tratamientos, se hace necesario difundir las características de presentación, pronósticas y evolutivas y el tratamiento de los pacientes con AC.
El objetivo del presente estudio es analizar las características de los pacientes con AC diagnosticados y seguidos en un centro de referencia español durante un periodo de 10 años.
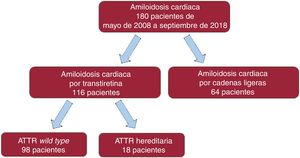
MÉTODOSSe llevó a cabo un estudio descriptivo, observacional y retrospectivo de todos los pacientes consecutivos diagnosticados de AC en el Hospital Universitario Puerta de Hierro Majadahonda desde mayo de 2008 hasta septiembre de 2018 (figura 1). El estudio fue aprobado por el Comité de ética del centro.

Se incluyó a pacientes con diagnóstico tanto invasivo como no invasivo. El diagnóstico invasivo se llevó a cabo mediante la demostración histológica de amiloide en una biopsia endomiocárdica (BEM) o en biopsias extracardiacas cuando se hallaba hipertrofia ventricular izquierda (HVI) (≥ 12mm) por ecocardiograma o resonancia magnética tras haber excluido otras causas de HVI12. Para la diferenciación entre AL y ATTR, se realizó inmunohistoquímica mediante anticuerpos monoclonales anti-AA, anti-TTR y anticadenas ligeras (DAKO, Dinamarca). En casos dudosos o con tinción por varios anticuerpos, se realizó espectrometría de masas para el diagnóstico definitivo del subtipo de amiloide.
El diagnóstico no invasivo de ATTR se estableció según los criterios publicados13 en presencia de datos compatibles con AC mediante técnicas de imagen cardiaca, una puntuación de Perugini de 2 o 3 en gammagrafía con 99mTc-DPD4 y habiendo excluido la AL mediante análisis de cadenas ligeras en suero (Freelite, The Binding Site Group, Reino Unido) e inmunofijación en sangre y orina. En todas las ATTR se realizó estudio genético mediante secuenciación completa del gen TTR.
En cuanto a los resultados hematológicos, se estableció el diagnóstico de gammapatía monoclonal de significado incierto o discrasia de células plasmáticas incluyendo el diagnóstico de mieloma múltiple así como la necesidad de biopsia de médula ósea según criterios vigentes14 tras valoración por hematología.
No se incluyó en el estudio a pacientes portadores de mutaciones en el gen TTR o con AL que no presentaban afección cardiaca.
VariablesLa información se obtuvo mediante revisión de los registros clínicos. Se obtuvieron datos de la primera evaluación y del seguimiento.
La información sobre el estado final del paciente se obtuvo de la historia clínica hospitalaria o de atención primaria. La mortalidad total se definió por muerte por cualquier causa a lo largo del seguimiento.
Análisis estadísticoLa normalidad de las variables se evaluó mediante el test de Shapiro-Wilk. Las variables continuas se expresan con media±desviación estándar para variables normales. Las variables categóricas se presentan mediante porcentajes. Para comparar las variables continuas, se utilizó la prueba de la t de Student bilateral para muestras independientes y para las categóricas, la prueba de la χ2 o el test de Fisher. Para calcular la supervivencia se utilizó la curva de Kaplan-Meier. Se consideró un valor p <0,05 como estadísticamente significativo.
Se realizó un análisis del tiempo hasta la muerte o el trasplante cardiaco entre los pacientes con AL y con ATTR. Además, se realizó un análisis multivariante de predictores de supervivencia mediante un modelo ajustado por edad, sexo, tipo de AC y las variables con p <0,10 en el modelo univariable (): insuficiencia cardiaca (IC) al diagnóstico, clase funcional de la New York Association (NYHA) III-IV al diagnóstico, fragmento aminoterminal del péptido natriurético cerebral (NT-proBNP)> 3.000 ng/dl, tasa de filtrado glomerular (continuo y <45ml/min/1,73 m2) y fracción de eyección del ventrículo izquierdo (FEVI) <50%.
Para el análisis estadístico se empleó STATA versión 12.0 StataCorp LP (Estados Unidos).
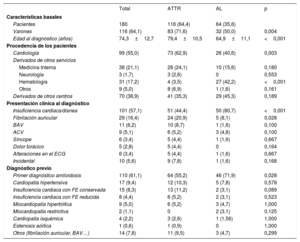
RESULTADOSSe incluyó a 180 pacientes con AC (el 64,1% varones; media de edad al diagnóstico, 74,3±12,7 años). De ellos, se diagnosticó AL a 64 (35,6%; el 50% varones; edad, 64,9±11,1 años) y ATTR a 116 (64,4%; el 71,6% varones; edad, 79,4±10,5 años). De estos, 98 (el 77% varones; edad, 80,8±8,5 años) eran pacientes con ATTRwt y 18 (el 44% varones; edad, 68,9±14,6 años) padecían ATTRh (figura 1). En la tabla 1 se exponen las características basales de todos los pacientes.
Perfil clínico al diagnóstico y diagnóstico previo
| Total | ATTR | AL | p | |
|---|---|---|---|---|
| Características basales | ||||
| Pacientes | 180 | 116 (64,4) | 64 (35,6) | |
| Varones | 116 (64,1) | 83 (71,6) | 32 (50,0) | 0,004 |
| Edad al diagnóstico (años) | 74,3±12,7 | 79,4±10,5 | 64,9±11,1 | <0,001 |
| Procedencia de los pacientes | ||||
| Cardiología | 99 (55,0) | 73 (62,9) | 26 (40,6) | 0,003 |
| Derivados de otros servicios | ||||
| Medicina Interna | 38 (21,1) | 28 (24,1) | 10 (15,6) | 0,180 |
| Neurología | 3 (1,7) | 3 (2,6) | 0 | 0,553 |
| Hematología | 31 (17,2) | 4 (3,5) | 27 (42,2) | <0,001 |
| Otros | 9 (5,0) | 8 (6,9) | 1 (1,6) | 0,161 |
| Derivados de otros centros | 70 (38,9) | 41 (35,3) | 29 (45,3) | 0,189 |
| Presentación clínica al diagnóstico | ||||
| Insuficiencia cardiaca/disnea | 101 (57,1) | 51 (44,4) | 50 (80,7) | <0,001 |
| Fibrilación auricular | 29 (16,4) | 24 (20,9) | 5 (8,1) | 0,028 |
| BAV | 11 (6,2) | 10 (8,7) | 1 (1,6) | 0,100 |
| ACV | 9 (5,1) | 6 (5,2) | 3 (4,8) | 0,100 |
| Síncope | 6 (3,4) | 5 (4,4) | 1 (1,6) | 0,667 |
| Dolor torácico | 5 (2,8) | 5 (4,4) | 0 | 0,164 |
| Alteraciones en el ECG | 6 (3,4) | 5 (4,4) | 1 (1,6) | 0,667 |
| Incidental | 10 (5,6) | 9 (7,8) | 1 (1,6) | 0,168 |
| Diagnóstico previo | ||||
| Primer diagnóstico amiloidosis | 110 (61,1) | 64 (55,2) | 46 (71,9) | 0,028 |
| Cardiopatía hipertensiva | 17 (9,4) | 12 (10,3) | 5 (7,8) | 0,578 |
| Insuficiencia cardiaca con FE conservada | 15 (8,3) | 13 (11,2) | 2 (3,1) | 0,089 |
| Insuficiencia cardiaca con FE reducida | 8 (4,4) | 6 (5,2) | 2 (3,1) | 0,523 |
| Miocardiopatía hipertrófica | 9 (5,0) | 6 (5,2) | 3 (4,7) | 1,000 |
| Miocardiopatía restrictiva | 2 (1,1) | 0 | 2 (3,1) | 0,125 |
| Cardiopatía isquémica | 4 (2,2) | 3 (2,6) | 1 (1,56) | 1,000 |
| Estenosis aórtica | 1 (0,6) | 1 (0,9) | 0 | 1,000 |
| Otros (fibrilación auricular, BAV…) | 14 (7,8) | 11 (9,5) | 3 (4,7) | 0,299 |
ACV: accidente cerebrovascular; AL: amiloidosis por cadenas ligeras; ATTR: amiloidosis transtiretina; BAV: bloqueo auriculoventricular; ECG: electrocardiograma; FE: fracción de eyección.
Los valores expresan n (%) o media±desviación estándar.
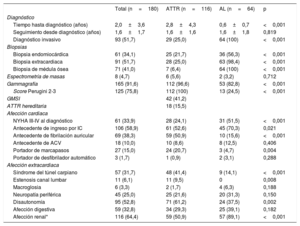
Entre mayo de 2008 y septiembre de 2018, se realizaron en total 223 biopsias: 61 (34%) BEM, 71 (41%) de médula ósea y 91 (51%) de otros órganos. Las BEM se realizaron mayoritariamente en el ventrículo derecho (56; 91,8%) y solo en 5 casos (8,2%) se obtuvieron muestras de VI. La tasa de complicaciones fue baja considerando que se trata de una técnica invasiva, con solo 2 complicaciones (3,3%) y en ambos casos no mortales. Las complicaciones ocurrieron tras BEM de ventrículo derecho y consistieron en un caso de arritmias ventriculares en el procedimiento y un derrame leve con taponamiento diferido en tratamiento anticoagulante. La rentabilidad de la BEM fue del 98,4%, y estableció el diagnóstico en el 56% (36) de las AL. Solo en un caso se realizó BEM del VI tras no demostrarse amiloide en la muestra de ventrículo derecho. En cuanto a biopsias extracardiacas, se realizaron 16 biopsias de glándula salival, 20 de grasa abdominal, 8 digestivas, 6 renales y 1 de nervio periférico, con rentabilidades variables y bajas, a excepción de las biopsias renal y de nervio periférico, dirigidas al órgano afectado (el 19, el 40, el 60, el 80 y el 100% respectivamente). En las biopsias de médula ósea realizadas a pacientes con sospecha de AL, se constató la presencia de más de un 10% de células plasmáticas en el 56% de los casos (36 pacientes) y la presencia de amiloide en un 81% de los casos (52 pacientes).
El diagnóstico definitivo del subtipo de amiloidosis se obtuvo mediante espectrometría de masas en 8 casos (4,7%). La principal causa que motivó el empleo de esta técnica fue una doble tinción tanto para TTR como para una de las cadenas ligeras por inmunohistoquímica. Los resultados de proteómica catalogaron 2 de los casos como AL y 6 como ATTR, coincidiendo con la sospecha clínica inicial en todos los casos salvo en uno.
El diagnóstico de AC ATTR se estableció de forma no invasiva en 87 pacientes (75%). La realización de BEM solo fue necesaria en 25 de ellos (22%). Se diagnosticó a 4 pacientes en presencia de afección cardiaca y biopsia extracardiaca positiva (3 de grasa abdominal y 1 digestiva).
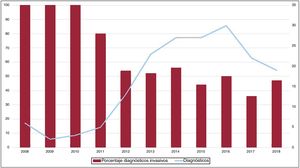
Aunque el porcentaje de pacientes diagnosticados de manera no invasiva en el total de la serie ha aumentado en los últimos años, el número de pacientes diagnosticados por biopsia sigue siendo relevante (figura 2).
 diagnosticados de forma invasiva y número total de pacientes con AC diagnosticados por año.")
Se analizó el gen TTR de todos sujetos diagnosticados de AC por ATTR y se encontró un total de 18 pacientes (16%) portadores de las siguientes mutaciones: 6 portadores de Val50Met, 6 Val142Ile, 3 Glu109Lys, 1 Glu109Gln, 1 Thr80Ala y 1 Ala65Thr. De los pacientes diagnosticados de ATTRh, 14 (78%) fueron los primeros individuos diagnosticados de sus familias. Entre los 114 pacientes con ATTR mayores de 60 años estudiados, se documentó una mutación en un 12% (14 casos). Estos resultados permitieron el estudio de 57 familiares de primer grado pertenecientes a 16 familias, y se encontró a otros 20 individuos (35%) portadores de mutaciones en el gen TTR.
Se realizó una gammagrafía con 99Tc-DPD a 165 pacientes (92%). De ellos, la mayoría (112; 68%) eran pacientes con ATTR que presentaron, principalmente (91%), una puntuación de Perugini de 3. Entre los 53 pacientes con AL a los que se realizó una gammagrafía con 99Tc-DPD, el 32% (17) presentaron algún grado de captación. Tenían una captación de grado 3 7 pacientes; de grado 2, 6 pacientes, y de grado 1, 4 casos. De los 13 pacientes con AL y captación de grados 2-3, 7 tenían captación únicamente en el VI; 5, en ambos ventrículos, y 1 solo en el ventrículo derecho.
El tiempo desde el inicio de los síntomas hasta el diagnóstico definitivo fue significativamente mayor en el grupo de ATTR, con una media de 2,8±4,3 frente a 0,6±0,7 años con AL (p <0,001); 70 pacientes (40%) ya tenían un diagnóstico establecido de una afección cardiaca distinta (tabla 1). El diagnóstico previo se produjo en el 45 y el 28% de los pacientes con ATTR y AL respectivamente. El diagnóstico previo más frecuente en ambos subtipos en conjunto fue la cardiopatía hipertensiva (17 pacientes; 9,4%), seguida por la IC con FEVI conservada, que fue especialmente frecuente entre los pacientes con ATTR (12; 10%).
Origen de los pacientes y presentación clínicaEl grueso de los pacientes diagnosticados con AC procedía de Cardiología (99; 55%), seguido de Hematología (31 pacientes, 17%), de donde procedían el 42% de las formas de AL, pero también el 4% de las ATTR. Entre los pacientes con ATTR, hasta un 41% cumplía criterios de gammapatía monoclonal de significado incierto.
Aunque en nuestro centro hay especial interés en esta enfermedad y desde hace unos años se plantea su diagnóstico de manera sistemática para los pacientes ingresados con IC que muestran aumento de grosores ventriculares, más de un tercio de los pacientes (70; 39%) fueron derivados desde otros centros (tabla 1). Proporcionalmente, fueron más frecuentes las derivaciones de pacientes con AL (45%) que con ATTR (35%).
En cuanto a la forma de presentación clínica, la IC/disnea fue la más frecuente y la presentación inicial en el 81% de las AL, frente a solo el 44% de las ATTR (p <0,001). A diferencia de las AL, la presentación clínica que motivó el diagnóstico fue mucho más variada en la ATTR, desde la fibrilación auricular (el 21% en ATTR frente al 8% en AL; p=0,028) y los trastornos de conducción (6%) a las alteraciones electrocardiográficas (3%), como parte del estudio etiológico de causas de ictus (5%) o incidentalmente tras la realización de una gammagrafía por motivos reumatológicos u oncológicos (6%).
El diagnóstico de AC se realizó durante un ingreso en el 53% de los casos (el 69% en AL, el 37% en ATTRh y el 45% en ATTRwt).
Afección cardiacaHasta un tercio de los pacientes (61; 34%) se encontraban en NYHA III-IV en el momento del diagnóstico. Más de la mitad (106; 59%) ya había requerido algún ingreso por IC, especialmente el subgrupo AL (el 70 frente al 53%; p=0,021). Sin embargo, se objetivó un mayor porcentaje de pacientes con antecedentes de fibrilación auricular (el 50 frente al 16%; p <0,001) y una mayor tasa de marcapasos en la primera evaluación (el 21 frente al 5%; p=0,004) entre los ATTR. Tres pacientes (2%) eran portadores de desfibrilador automático implantable en la primera evaluación.
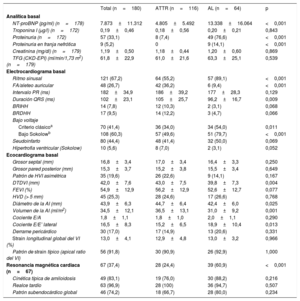
En cuanto a biomarcadores, el NT-proBNP medio fue de 7.873±11.312 pg/ml, significativamente más alto en AL que en ATTR (13.338±16.064 frente a 4.805±5.492 pg/ml; p <0,001) y entre pacientes diagnosticados durante un ingreso con respecto a los diagnosticados sin estar ingresados (10.987±13.704 frente a 4.308±6.068 pg/ml; p <0,001). No hubo diferencias significativas en troponina I entre AL y ATTR (0,18±0,56 frente a 0,20±0,21μg/l; p=0,843). Tampoco hubo diferencias en las tasas de filtrado glomerular, aunque los pacientes con AL presentaron con más frecuencia proteinuria en la franja nefrótica (tabla 2).
Diagnóstico y características clínicas de los pacientes con amiloidosis cardiaca según subtipo
| Total (n=180) | ATTR (n=116) | AL (n=64) | p | |
|---|---|---|---|---|
| Diagnóstico | ||||
| Tiempo hasta diagnóstico (años) | 2,0±3,6 | 2,8±4,3 | 0,6±0,7 | <0,001 |
| Seguimiento desde diagnóstico (años) | 1,6±1,7 | 1,6±1,6 | 1,6±1,8 | 0,819 |
| Diagnóstico invasivo | 93 (51,7) | 29 (25,0) | 64 (100) | <0,001 |
| Biopsias | ||||
| Biopsia endomiocárdica | 61 (34,1) | 25 (21,7) | 36 (56,3) | <0,001 |
| Biopsia extracardiaca | 91 (51,7) | 28 (25,0) | 63 (98,4) | <0,001 |
| Biopsia de médula ósea | 71 (41,0) | 7 (6,4) | 64 (100) | <0,001 |
| Espectrometría de masas | 8 (4,7) | 6 (5,6) | 2 (3,2) | 0,712 |
| Gammagrafía | 165 (91,6) | 112 (96,6) | 53 (82,8) | <0,001 |
| Score Perugini 2-3 | 125 (75,8) | 112 (100) | 13 (24,5) | <0,001 |
| GMSI | 42 (41,2) | |||
| ATTR hereditaria | 18 (15,5) | |||
| Afección cardiaca | ||||
| NYHA III-IV al diagnóstico | 61 (33,9) | 28 (24,1) | 31 (51,5) | <0,001 |
| Antecedente de ingreso por IC | 106 (58,9) | 61 (52,6) | 45 (70,3) | 0,021 |
| Antecedente de fibrilación auricular | 69 (38,3) | 59 (50,9) | 10 (15,6) | <0,001 |
| Antecedente de ACV | 18 (10,0) | 10 (8,6) | 8 (12,5) | 0,406 |
| Portador de marcapasos | 27 (15,0) | 24 (20,7) | 3 (4,7) | 0,004 |
| Portador de desfibrilador automático | 3 (1,7) | 1 (0,9) | 2 (3,1) | 0,288 |
| Afección extracardiaca | ||||
| Síndrome del túnel carpiano | 57 (31,7) | 48 (41,4) | 9 (14,1) | <0,001 |
| Estenosis canal lumbar | 11 (6,1) | 11 (9,5) | 0 | 0,008 |
| Macroglosia | 6 (3,3) | 2 (1,7) | 4 (6,3) | 0,188 |
| Neuropatía periférica | 45 (25,0) | 25 (21,6) | 20 (31,3) | 0,150 |
| Disautonomía | 95 (52,8) | 71 (61,2) | 24 (37,5) | 0,002 |
| Afección digestiva | 59 (32,8) | 34 (29,3) | 25 (39,1) | 0,182 |
| Afección renal* | 116 (64,4) | 59 (50,9) | 57 (89,1) | <0,001 |
ACV: accidente cerebrovascular; AL: amiloidosis por cadenas ligeras; ATTR: amiloidosis transtiretina; GMSI: gammapatía monoclonal de significado incierto; NYHA: clase funcional de la New York Heart Association.
Los valores expresan n (%) o media±desviación estándar.
Entre los pacientes con AL, 11 (17%) se encontraban en estadio Mayo III al diagnóstico y 48 (75%), en estadio IV. En los ATTR, 39 (34%) se encontraban en estadio II y 25 (22%) en estadio III de Gillmore15.
En los electrocardiogramas, una cuarta parte de la serie presentaba fibrilación auricular en la primera evaluación, significativamente más frecuente en ATTR (el 36 frente al 9%; p <0,001). En las formas AL, el clásico patrón de bajos voltajes se halló en 34 pacientes (54%) y el patrón de seudoinfarto, en 32 (50%), que constituyó el patrón electrocardiográfico más frecuente en toda la cohorte (tabla 3).
Pruebas complementarias basales en pacientes con amiloidosis cardiaca según subtipo
| Total (n=180) | ATTR (n=116) | AL (n=64) | p | |
|---|---|---|---|---|
| Analítica basal | ||||
| NT-proBNP (pg/ml) (n=178) | 7.873±11.312 | 4.805±5.492 | 13.338±16.064 | <0,001 |
| Troponina I (μg/l) (n=172) | 0,19±0,46 | 0,18±0,56 | 0,20±0,21 | 0,843 |
| Proteinuria (n=172) | 57 (33,1) | 8 (7,4) | 49 (76,6) | <0,001 |
| Proteinuria en franja nefrótica | 9 (5,2) | 0 | 9 (14,1) | <0,001 |
| Creatinina (mg/dl) (n=179) | 1,19±0,50 | 1,18±0,44 | 1,20±0,60 | 0,869 |
| TFG (CKD-EPI) (ml/min/1,73 m2) (n=179) | 61,8±22,9 | 61,0±21,6 | 63,3±25,1 | 0,539 |
| Electrocardiograma basal | ||||
| Ritmo sinusal | 121 (67,2) | 64 (55,2) | 57 (89,1) | <0,001 |
| FA/aleteo auricular | 48 (26,7) | 42 (36,2) | 6 (9,4) | <0,001 |
| Intervalo PR (ms) | 182±34,9 | 186±39,2 | 177±28,3 | 0,129 |
| Duración QRS (ms) | 102±23,1 | 105±25,7 | 96,2±16,7 | 0,009 |
| BRIHH | 14 (7,8) | 12 (10,3) | 2 (3,1) | 0,068 |
| BRDHH | 17 (9,5) | 14 (12,2) | 3 (4,7) | 0,066 |
| Bajo voltaje | ||||
| Criterio clásicoa | 70 (41,4) | 36 (34,0) | 34 (54,0) | 0,011 |
| Bajo Sokolowb | 108 (60,3) | 57 (49,6) | 51 (79,7) | <0,001 |
| Seudoinfarto | 80 (44,4) | 48 (41,4) | 32 (50,0) | 0,069 |
| Hipertrofia ventricular (Sokolow) | 10 (5,6) | 8 (7,0) | 2 (3,1) | 0,052 |
| Ecocardiograma basal | ||||
| Grosor septal (mm) | 16,8±3,4 | 17,0±3,4 | 16,4±3,3 | 0,250 |
| Grosor pared posterior (mm) | 15,3±3,7 | 15,2±3,8 | 15,5±3,4 | 0,649 |
| Patrón de HVI asimétrica | 35 (19,6) | 26 (22,6) | 9 (14,1) | 0,167 |
| DTDVI (mm) | 42,0±7,6 | 43,0±7,5 | 39,8±7,3 | 0,004 |
| FEVI (%) | 54,9±12,9 | 56,2±12,9 | 52,6±12,7 | 0,077 |
| HVD (> 5 mm) | 45 (25,3) | 28 (24,6) | 17 (26,6) | 0,768 |
| Diámetro de la AI (mm) | 43,9±6,3 | 44,7±6,4 | 42,4±6,0 | 0,025 |
| Volumen de la AI (ml/m2) | 34,5±12,1 | 36,5±13,1 | 31,0±9,2 | 0,001 |
| Cociente E/A | 1,8±1,1 | 1,8±1,0 | 2,0±1,1 | 0,290 |
| Cociente E/E’ lateral | 16,5±8,3 | 15,2±6,5 | 18,9±10,4 | 0,013 |
| Derrame pericárdico | 30 (17,0) | 17 (14,9) | 13 (20,6) | 0,331 |
| Strain longitudinal global del VI (%) | 13,0±4,1 | 12,9±4,8 | 13,0±3,2 | 0,966 |
| Patrón de strain típico (apical ratio del VI) | 56 (91,8) | 30 (90,9) | 26 (92,9) | 1,000 |
| Resonancia magnética cardiaca (n=67) | 67 (37,4) | 28 (24,4) | 39 (60,9) | <0,001 |
| Cinética típica de amiloidosis | 49 (83,1) | 19 (76,0) | 30 (88,2) | 0,216 |
| Realce tardío | 63 (96,9) | 28 (100) | 36 (94,7) | 0,507 |
| Patrón subendocárdico global | 46 (74,2) | 18 (66,7) | 28 (80,0) | 0,234 |
AI: aurícula izquierda; AL: amiloidosis por cadenas ligeras; ATTR: amiloidosis por transtiretina; BRDHH: bloqueo de rama derecha del haz de His; BRIHH: bloqueo de rama izquierda del haz de His; CKD-EPI: Chronic Kidney Disease Epidemiology Collaboration; DTDVI: diámetro telediastólico del ventrículo izquierdo; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; HVD: hipertrofia del ventrículo derecho; HVI: hipertrofia ventricular izquierda; NT-proBNP: fracción aminoterminal del propéptido natriurético cerebral; TFG: tasa de filtrado glomerular; VI: ventrículo izquierdo.
Los valores expresan n (%) o media ± desviación estándar.
En cuanto a la valoración ecocardiográfica, el grosor septal medio de la cohorte fue de 16,8±3,4mm, con un 20% de pacientes con HVI excéntrica. No se observaron diferencias significativas entre ATTR y AL en cuanto a la FEVI, si bien hubo cierta tendencia a peor FEVI en la AL (el 56,2±12,9% frente al 52,6±12,7%; p=0,077). En contraposición, las formas de AL presentaron menor dilatación auricular izquierda (44,7±6,4 frente a 42,4±6,0mm; p=0,025; 36,5±13,1 frente a 31,0±9,2ml/m2; p=0,001), pero con cociente E/E’ lateral significativamente mayor (15,2±6,5 frente a 18,9±10,4cm/s; p=0,013).
De los 67 pacientes a los que se realizó una resonancia cardiaca (37%), en más del 80% se observó el comportamiento característico de cinética acelerada de gadolinio con anulación del miocardio antes de la sangre. Además, en la práctica totalidad se evidenció realce tardío (63; 96,9%), con un patrón subendocárdico global en más de la mitad de los pacientes (18; 67%) y especialmente en las formas de AL (28; 80%).
Durante el seguimiento, 9 pacientes (6%) requirieron implante de marcapasos (6 con ATTR y 3 con AL) y en 2 pacientes se implantó un desfibrilador en prevención primaria (uno a 1 paciente con AL y fenotipo restrictivo al que se implantó un marcapasos resincronizador en otro centro tras documentarse un bloqueo completo, y otro a 1 paciente con ATTRwt con disfunción ventricular grave y síncope).
Afección extracardiacaHabía datos clínicos de depósito de amiloide en el tejido conectivo en más de la mitad de los pacientes con ATTR, principalmente en forma de síndrome del túnel carpiano (48; 41%), seguido de estenosis del canal lumbar (11; 10%). También las formas de AL presentaban síndrome del túnel carpiano, aunque con menos frecuencia (14%; p <0,001). Los síntomas neurológicos y digestivos (vómitos o diarrea) fueron relativamente frecuentes (el 25 y el 33% respectivamente). Presentaban afección neurológica 13 de los 18 pacientes con ATTRh (72%) y también en un grupo de las ATTRwt se describían síntomas de neuropatía periférica (12%).
Tratamiento dirigido y trasplanteDurante el periodo del estudio, 18 pacientes (10%) participaron en algún ensayo clínico con moléculas antiamiloideas específicas.
En este periodo, 15 pacientes con ATTR (13%) recibieron algún tratamiento dirigido. La mayoría (12) recibió tratamiento estabilizador (tafamidis en 10 casos y diflunisal en 2). Se trató a 3 pacientes con ATTRh y afección neurológica con un silenciador genético (patisirán) destinado a suprimir la producción de TTR tras la progresión de la enfermedad en tratamiento estabilizador (2) o directamente de inicio (1). Por último, 2 pacientes siguieron tratamiento con epigalocatequina-3 galato, polifenol del té verde.
En cuanto a los pacientes con AL, la mayoría (72%) recibió tratamiento antineoplásico, 44 con combinaciones de bortezomib y 2 con alquilantes. Se sometió a 6 pacientes a trasplante autólogo de progenitores hematopoyéticos tras otro tratamiento previo, y 18 no llegaron a recibir tratamiento o recibieron un único ciclo por fallecimiento precoz.
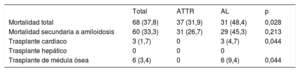
Durante el periodo analizado, ningún paciente con ATTRh recibió un trasplante hepático. En cambio, 3 pacientes con AL se sometieron a trasplante cardiaco (tabla 4).
Mortalidad y trasplante en pacientes con amiloidosis cardiaca según el subtipo
| Total | ATTR | AL | p | |
|---|---|---|---|---|
| Mortalidad total | 68 (37,8) | 37 (31,9) | 31 (48,4) | 0,028 |
| Mortalidad secundaria a amiloidosis | 60 (33,3) | 31 (26,7) | 29 (45,3) | 0,213 |
| Trasplante cardiaco | 3 (1,7) | 0 | 3 (4,7) | 0,044 |
| Trasplante hepático | 0 | 0 | 0 | |
| Trasplante de médula ósea | 6 (3,4) | 0 | 6 (9,4) | 0,044 |
AL: amiloidosis por cadenas ligeras; ATTR: amiloidosis por transtiretina.
Durante una media de seguimiento de 1,6±1,7 años, la mortalidad total de la cohorte fue del 38% (68 pacientes), significativamente mayor en AL (el 48%, frente al 32% en ATTR; p=0,028) (tabla 4). En 60 de los fallecidos (88%) la causa de la muerte puede atribuirse a la amiloidosis.
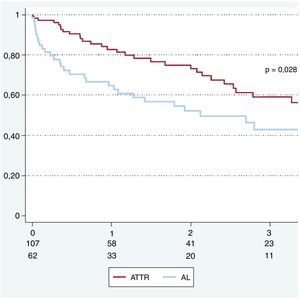
La supervivencia total al año fue del 76%. La supervivencia a 12, 24 y 36 meses en AL fue del 65, el 52 y el 43% y en ATTR, del 83, el 73 y el 59% respectivamente (figura 3).

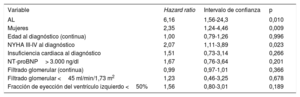
En el análisis multivariable, los únicos factores asociados de manera significativa con muerte o trasplante cardiaco fueron el subtipo de AL (hazard ratio [HR]=6,16; intervalo de confianza del 95% [IC95%], 1,56-24,3; p=0,010), el sexo femenino (HR=2,35; IC95%, 1,24-4,46; p=0,009] y la NYHA III-IV al diagnóstico (HR=2,07; IC95%, 1,11-3,89; p=0,023) (tabla 5).
Análisis multivariable de los factores predictores de mortalidad en 180 pacientes con amiloidosis cardiaca
| Variable | Hazard ratio | Intervalo de confianza | p |
|---|---|---|---|
| AL | 6,16 | 1,56-24,3 | 0,010 |
| Mujeres | 2,35 | 1,24-4,46 | 0,009 |
| Edad al diagnóstico (continua) | 1,00 | 0,79-1,26 | 0,996 |
| NYHA III-IV al diagnóstico | 2,07 | 1,11-3,89 | 0,023 |
| Insuficiencia cardiaca al diagnóstico | 1,51 | 0,73-3,14 | 0,266 |
| NT-proBNP> 3.000 ng/dl | 1,67 | 0,76-3,64 | 0,201 |
| Filtrado glomerular (continua) | 0,99 | 0,97-1,01 | 0,366 |
| Filtrado glomerular <45 ml/min/1,73 m2 | 1,23 | 0,46-3,25 | 0,678 |
| Fracción de eyección del ventrículo izquierdo <50% | 1,56 | 0,80-3,01 | 0,189 |
AL: amiloidosis por cadenas ligeras; NT-proBNP: fracción aminoterminal del propéptido natriurético cerebral; NYHA: clase funcional de la New York Heart Association.
Este estudio describe las características de una cohorte amplia de pacientes con AC. Mediante el análisis de la práctica clínica de un centro con experiencia en el tratamiento y el seguimiento de esta entidad, se muestra que la ATTR es la forma más frecuente de AC en nuestro medio. Quedan de manifiesto las variadas formas de presentación de la AC, así como las heterogéneas manifestaciones electrocardiográficas y ecocardiográficas. Nuestro trabajo pone de manifiesto también la alta rentabilidad de la BEM en las formas de AL y que en los últimos años ha crecido el número de pacientes con ATTR diagnosticados de manera no invasiva. Por último, se identifica el subtipo de AL, la clase funcional avanzada y el sexo femenino como factores predictores de mortalidad o trasplante.
Hasta la fecha, son pocas las series publicadas que describan las características de pacientes con AC16,17. En los últimos años, hemos asistido a un creciente interés por la AC, con la aparición de un gran número de estudios en este campo. Nuestros datos son fruto de la experiencia de un centro con programa de trasplante cardiaco y una unidad de cardiopatías familiares, pioneros en la atención a estos enfermos, así como el resultado del aprendizaje continuo sobre la enfermedad1,2,18-25.
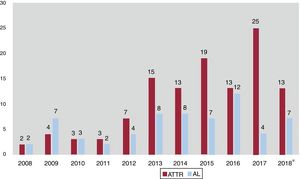
Hasta ahora, la AL se consideraba la forma más frecuente de AC en países desarrollados, y la ATTR es un subgrupo poco representado en las publicaciones previas. Sin embargo, los datos actuales apuntan a que la ATTR es en realidad la AC más frecuente5 y hasta ahora ha estado infradiagnosticada, con una prevalencia real no conocida. La ATTR constituye hoy más de la mitad de los pacientes atendidos en nuestro centro y asistimos a un cambio de tendencia, con un claro aumento de estos pacientes en los últimos años (figura 4). La posibilidad de disponer de tratamientos específicos para el tratamiento de la ATTR2 hace aún más indispensable identificar a estos pacientes y contribuirá probablemente a aumentar el número de pacientes diagnosticados de AC, tanto ATTR como AL.

El diagnóstico de AC sigue siendo un punto clave. Nuestros datos muestran que el cuadro clásico descrito (miocardiopatía restrictiva con hipertrofia concéntrica y bajos voltajes en ECG) solo está presente en una minoría de los pacientes. Aunque la IC es la manifestación más frecuente, a diferencia de las formas de AL, el perfil clínico de la ATTR es mucho más variado. Este hallazgo requiere un cambio de mentalidad a la hora de sospechar esta afección. El tiempo medio desde el inicio de los síntomas hasta el diagnóstico en los pacientes atendidos en nuestro centro fue 0,6±0,7 años en AL y 2,8±4,3 años en ATTR. El retraso diagnóstico medio en ambos grupos es inferior al descrito en la literatura, un retraso de alrededor de 3 años en las formas ATTR y menos de la mitad de los pacientes con AL diagnosticados en los primeros 6 meses6. Creemos que el menor tiempo observado en nuestros pacientes es consecuencia de una alta sospecha clínica, la realización precoz de BEM y el elevado número de pacientes con ATTR con diagnóstico no invasivo.
La experiencia acumulada en la realización de más de 20.000 gammagrafías óseas con 99Tc-DPD a lo largo de los últimos 10 años avala, en nuestra opinión, la incorporación sistemática de esta prueba en el proceso diagnóstico de AC por ATTR. De hecho, se diagnosticó de manera no invasiva al 72% de los pacientes con ATTR en nuestra serie. También la resonancia, con su gran capacidad para la caracterización morfológica, funcional y tisular, es fundamental en el diagnóstico no invasivo de la AC19. La posibilidad de evaluar parámetros como el volumen extracelular y el mapeo en T1 probablemente incremente su rendimiento diagnóstico en los próximos años26.
Sin embargo, la demostración histológica sigue siendo fundamental en un grupo relevante de pacientes y particularmente en aquellos con sospecha de AL. Nuestro grupo ya describió la importancia de dirigirse al órgano afectado sin demora8 y los datos actuales muestran la alta rentabilidad de la BEM.
Una vez establecido el diagnóstico de AC, es crítico determinar el subtipo. Esta distinción no siempre es sencilla. Así, no toda captación en la gammagrafía es equivalente a ATTR y no todo componente monoclonal en AC implica el tipo AL. Nosotros describimos una prevalencia significativa de gammapatía monoclonal de significado incierto en la población ATTR (41%), superior a cohortes previas y mayor respecto a la población general de su misma edad. En este contexto, la utilización de técnicas inmunohistoquímicas y análisis proteómico es fundamental. En nuestro caso, la espectrometría de masas fue necesaria para determinar el subtipo de amiloidosis en 5% de los pacientes biopsiados.
Finalmente, nuestros datos muestran una supervivencia del 76% a 1 año, equiparable a la de otros centros de referencia16, y con peor pronóstico en las formas de AL frente a las ATTR (supervivencia del 65 frente al 83% a 1 año). Existe un consenso generalizado sobre la utilidad de los nuevos fármacos antimieloma (inhibidores del proteasoma, inmunomoduladores y anticuerpos monoclonales) en el tratamiento de los pacientes con AL, y la utilización de dosis altas de quimioterapia y el trasplante autólogo permiten obtener remisiones de larga duración en pacientes seleccionados10. El mal pronóstico de los pacientes con AL y la complejidad de los tratamientos requeridos refuerzan la necesidad de que estos pacientes sean atendidos por equipos especializados que cuenten con la colaboración estrecha de unidades de IC avanzada y hematología.
A la luz de los resultados presentados, cabe señalar que la AC sigue siendo una entidad con mal pronóstico, incluso en centros de referencia. Creemos que la complejidad del diagnóstico y el tratamiento de los pacientes con ATTR y AL, así como la posibilidad de diversos tratamientos específicos, avalan la creación de centros de referencia que dispongan de los recursos y la experiencia para dar respuesta a las necesidades de estos pacientes, de modo análogo a otros países de nuestro entorno27.
LimitacionesSe trata de un estudio descriptivo y retrospectivo, realizado en un único centro y sujeto a sesgos de selección y supervivencia. Cabe resaltar que cerca del 80% de los diagnósticos se han realizado en los últimos 5 años, lo que también puede influir en los resultados.
CONCLUSIONESLa ATTR es hoy la forma más frecuente de AC. La presentación clínica y las características de los pacientes con AC es heterogénea y difiere entre la ATTR y la AL. La BEM presenta una alta rentabilidad diagnóstica con pocas complicaciones. Hoy se diagnostica de manera no invasiva a una parte importante de los pacientes con ATTR. El subtipo de AL, el sexo femenino y la clase NYHA III-IV al diagnóstico se asocian de manera independiente con peor pronóstico. En un contexto de aparición de nuevos tratamientos para la AC, es necesario que los cardiólogos se familiaricen con las características y el proceso diagnóstico de estos pacientes.
CONFLICTO DE INTERESESE. Gonzalez-Lopez ha recibido honorarios por ponencias y actividad de asesoría de Pfizer y Proclara. I. Krsnik ha recibido honorarios por ponencias de Janssen, por actividad de asesoría de Celgene y Prothena y por financiación para investigación de Prothena. P. Garcia-Pavia ha recibido honorarios por ponencias o actividad de asesoría de Akcea, Alnylam, Eidos, Neuroinmmune, Pfizer y Prothena, y financiación para investigación de Alnylam, Pfizer y Prothena.
- –
La AC es una entidad infradiagnosticada que presenta un considerable retraso diagnóstico.
- –
En los últimos años se han producido numerosos avances con la posibilidad de establecer el diagnóstico de manera no invasiva y el desarrollo de nuevas terapias.
- –
La ATTR es la forma más frecuente de AC en nuestro medio.
- –
La presentación clínica y las características de los pacientes con AC es heterogénea y difiere sustancialmente entre las formas ATTR y AL.
- –
En nuestro medio, el diagnóstico de AC por ATTR se realiza de manera no invasiva cada vez con mayor frecuencia.
- –
El subtipo de AL, el sexo femenino y la clase NYHA III-IV al diagnóstico se asocian de modo independiente con peor pronóstico.
A Ariadna González y Almudena Ariza, por su labor en la coordinación de pacientes y la gestión del portal Amiloidosises del Hospital Puerta de Hierro Majadahonda.
A los Dres. Lucía Galán y Antonio Guerrero del Servicio de Neurología del Hospital Clínico San Carlos, Madrid, por la atención de los pacientes con ATTRh.