Las plaquetas tienen un papel fundamental tanto en la hemostasia como en la patogenia de la aterotrombosis. Después de producirse la rotura de la placa de ateroma, diferentes proteínas se expresan en la plaqueta que interviene tanto en la unión de la plaqueta a la pared vascular dañada como en la interacción con nuevas plaquetas y otras células sanguíneas para formar el trombo final. Diferentes agonistas, entre ellos el difosfato de adenosina, el tromboxano A2 y la trombina, se sintetizan y se liberan para llamar a más plaquetas a formar parte del trombo. Además, diferentes inhibidores endógenos de las plaquetas tratan de formar los agonistas plaquetarios anteriormente mencionados. Las plaquetas jóvenes y las micropartículas derivadas de las plaquetas también participan en la formación del trombo. Este artículo trata de revisar los mecanismos fisiológicos implicados en la activación y la inactivación plaquetarias.
Palabras clave
La hemostasia es uno de los más importantes sistemas de defensa del organismo. En su inicio es probable que el sistema se desarrollara para evitar el sangrado; sin embargo, su desarrollo en entornos arteriales puede convertir el proceso en patológico.
Para que ocurra el proceso de activación de las plaquetas durante la aterotrombosis, se requiere una serie de condiciones: a) la existencia de disfunción endotelial; b) alteraciones en el metabolismo de lípidos y lipoproteínas; c) inflamación crónica en la que estén implicadas citocinas, quimiocinas, moléculas de adhesión y factores de crecimiento que modifican la funcionalidad de la pared vascular, intervienen en la regulación de la interacción intercelular y facilitan el desarrollo y la rotura de la placa ateromatosa; d) el estrés oxidativo que cambia la estructura y la actividad de lipoproteínas y promueve la activación de células, incluidas las propias plaquetas, y e) un estado hipercoagulante en el que las plaquetas también contribuyen.
Las plaquetas intervienen en el proceso trombótico agudo que sigue a la rotura de la placa de ateroma. Algunos datos experimentales también indican la posible participación de las plaquetas en la formación de la placa ateromatosa. Es evidente además que los tratamientos farmacológicos que permiten reducir el riesgo cardiovascular también reducen de manera indirecta, y algunos de ellos directamente, la activación de las plaquetas. Así, los tratamientos hipolipemiantes, antidiabéticos, antihipertensivos, vasodilatadores, etc., e incluso medidas dietéticas de mejora en los hábitos de vida pueden, en mayor o menor medida, reducir la activación de las plaquetas1–4. También existen fármacos desarrollados específicamente para inhibir la capacidad de activación de las plaquetas con papel terapéutico, tanto en prevención primaria, en pacientes con enfermedad aterosclerótica o factores de riesgo asociados, como en prevención secundaria, en pacientes que ya han sufrido un evento trombótico. Esta revisión intenta repasar los mecanismos de agregación y antiagregación plaquetaria que en cierta medida nos ayuden a comprender cómo funcionan los fármacos antiplaquetarios e identifiquen otras posibles dianas terapéuticas para el desarrollo de nuevos fármacos.
Las PlaquetasDe los elementos que forman la sangre, la plaqueta es el último en ser descubierto. Se considera al francés Alfied Donne (1801–1878) como el descubridor de las plaquetas, aunque también se atribuye al médico inglés George Gulliver (1804–1882). No fue hasta finales del siglo XIX cuando Giulio Bizzozero (1841–1901) aisló las plaquetas de los trombos e identificó la hemostasia y la trombosis como procesos análogos.
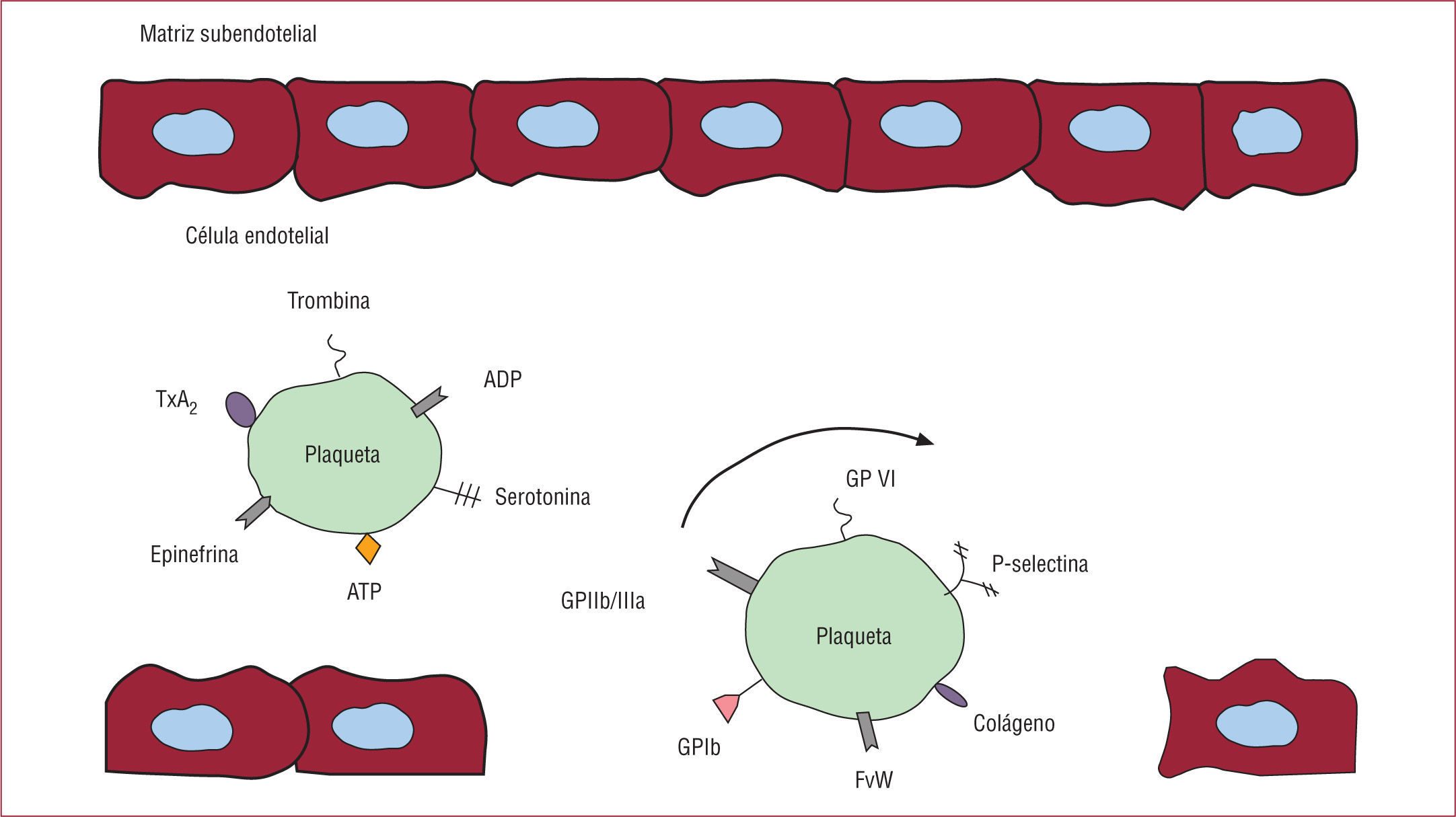
Las plaquetas son células enucleadas de 1–2μm de tamaño, generadas en la médula ósea por fragmentación de los bordes de los megacariocitos, que se acumulan en el lugar donde el endotelio está disfuncional o dañado dentro de la pared arterial, lo que inicia la formación del trombo5. El intervalo fisiológico de las plaquetas es de 150–400 × 109/l. Un adulto sano produce cada día una media de alrededor de 1×1011 plaquetas. La expectativa de vida de las plaquetas es de 7 a 10 días.
En condiciones fisiológicas, las plaquetas circulan en forma no activa y expresan en su superficie un número relativamente pequeño de muchas de las moléculas que, en estado activado, van a facilitar su interacción con otras plaquetas y otras células de su entorno. Además, las plaquetas contienen diferentes tipos de gránulos (fundamentalmente gránulos densos, gránulos α y lisosomas) desde los que al ser activadas liberan diferentes factores almacenados en ellos y que a su vez estimulan más la actividad de la propia plaqueta. Estos factores tienen también efectos biológicos sobre otras células del entorno plaquetario. Un estudio proteómico ha descrito que las plaquetas activadas por trombina liberan más de 300 proteínas diferentes, muchas de ellas relacionadas con reacciones inflamatorias6. Además, las plaquetas pueden interaccionar con patógenos bacterianos e incluso expresar receptores del complemento7, lo que las convierte también en células involucradas en la inmunogenicidad del organismo. De hecho, las plaquetas expresan y almacenan proteínas antibacterianas llamadas trombocidinas8.
Como hemos señalado, las plaquetas contienen fundamentalmente tres tipos de gránulos: los gránulos densos, los gránulos α y los lisosomas. La liberación de los gránulos densos en las plaquetas ocurre por exocitosis, y desde ellos se liberan difosfato de adenosina (ADP), trifosfato de adenosina (ATP), fosfato inorgánico, polifosfatos, serotonina y calcio, entre otros.
La liberación de ADP es esencial como cofactor de la agregación plaquetaria y actúa mediante su interacción con receptores específicos localizados en la superficie plaquetaria. Se conocen dos receptores para el ADP en la plaqueta, uno acoplado a la proteína Gq (el P2Y1) y otro acoplado a Gi (el P2Y12), que es esencial para la hemostasia primaria9,10. Ambos receptores actúan de modo sinérgico en la activación de las plaquetas. El P2Y1 probablemente sea lo que origina la activación inicial reversible, mientras que el P2Y12 es necesario para la activación prolongada y la agregación plaquetaria. El ADP y el ATP no sólo pueden actuar como coactivadores plaquetarios, sino también influir en el tono vascular.
El calcio liberado por la plaqueta es necesario para la formación de fibrina, mientras que los polifosfatos actúan como elementos reguladores en la coagulación y en el sistema fibrinolítico reaccionando con el factor XII11, entre otros. Finalmente, la serotonina no sólo tiene efecto vasoconstrictor, sino que también interviene en la activación de las propias plaquetas.
Los lisosomas plaquetarios contienen elastasas y otras proteasas que facilitan la degradación de la matriz extracelular, además de crear un ambiente ácido que favorecerá la acción de estas enzimas.
Los gránulos α son reservorios de proteínas que van desde factores de crecimiento hasta moléculas de adhesión o receptores que utiliza la plaqueta para interaccionar con otras células. Entre estos receptores se incluyen las glucoproteínas (GP) Ib y αIIbβ312. Otra de las moléculas de adhesión contenidas en estos gránulos es la P-selectina, que permite la interacción de las plaquetas con las células endoteliales, los leucocitos y otras células inmunitarias. En los gránulos α hay también moléculas asociadas a la respuesta inflamatoria, como las citocinas.
Activación PlaquetariaEl mecanismo de formación del trombo plaquetario puede dividirse en cuatro etapas:
- 1.
Frenado de las plaquetas circulantes sobre la pared vascular contra la corriente del flujo sanguíneo que las empuja.
- 2.
Activación y adhesión firme de la plaqueta a la pared del vaso.
- 3.
Unión de más plaquetas a las ya adheridas, que sería la fase de crecimiento del trombo.
- 4.
Estabilización del trombo, la última fase.
En cada fase actúa una serie de mecanismos no completamente conocidos.
La GPIbα actúa en la fase inicial de frenado de las plaquetas sobre la pared vascular. La GPIbα se expresa de forma constitutiva en la superficie de la plaqueta e inicia el proceso de adhesión plaquetario uniéndose al colágeno y al factor von Willebrand (FvW)13. El FvW estará embebido en las fibras de colágeno, particularmente del colágeno de tipos I, III y VI. En los vasos con alto estrés de rozamiento, como ocurre en las arterias, el FvW es esencial para reducir el flujo rápido de las plaquetas mediante la interacción del dominio A1 del FvW con GPIbα. Sin embargo, la GPIbα es también el receptor más conocido de la proteína Mac-1, localizada en la superficie de los leucocitos activados. Mediante la interacción entre GPIbα y Mac-1 ocurre la unión entre plaqueta y leucocito, importante en la respuesta inflamatoria mediada por las plaquetas.
La interacción transitoria entre el FvW y la GPIbα permite la «rodadura » de las plaquetas en la zona dañada del vaso. Como resultado, las proteínas contenidas en la pared vascular, fundamentalmente el colágeno, inducirán la activación de las plaquetas y su adhesión firme a la pared, de tal manera que el colágeno y el FvW forman una especie de unidad funcional para la formación inicial del trombo, en el que el FvW contribuye a la captura inicial de las plaquetas en la superficie del vaso y el colágeno permite que se establezca una unión más estable con las plaquetas. En el proceso de interacción entre plaqueta y colágeno participan dos receptores plaquetarios, la GPVI y la integrina α2β114.
La activación de las plaquetas mediada por GPVI permite una firme adhesión de las plaquetas y la secreción de las sustancias procoagulantes y proinflamatorias contenidas en ellas, lo que hace que el trombo crezca y se consolide su formación. Además, a la unión de las plaquetas al colágeno sigue la expresión de fosfatidilserina sobre la membrana plaquetaria. La fosfatidilserina proporciona actividad protrombinasa, que aumenta la formación de trombina. Las plaquetas adheridas permanecerán vivas durante horas o días en el sitio de la lesión vascular y liberarán microvesículas con actividad proinflamatoria y protrombótica, de las cuales se habla más adelante.
Después de la deposición de las plaquetas sobre el FvW y el colágeno, se requiere el reclutamiento de nuevas plaquetas desde la circulación, en un proceso conocido como agregación plaquetaria. Esto es posible por la acumulación local de agonistas de la activación de las plaquetas debida a su secreción desde las plaquetas ya adheridas a la pared del vaso. Entre estos agonistas se incluyen el ADP, el TxA2, la epinefrina y la trombina. La etapa final es la activación de los receptores αIIbβ3, que posibilitan la unión del fibrinógeno y también del FvW, lo que permite el establecimiento de puentes estables entre las plaquetas. En el proceso de estabilización participan también otras moléculas, quizá una de las de mayor interés sea el ligando de CD40 (CD40L).
El CD40L es una GP almacenada en los gránulos plaquetarios que, tras la desgranulación plaquetaria, pasa a expresarse en la superficie de la plaqueta. Desde allí puede liberarse desde la plaqueta al plasma mediante la actividad de la metaloproteasa-2. Tanto el CD40L unido a la plaqueta como el CD40L soluble interaccionan con el CD40, expresado en los linfocitos B, los neutrófilos, los monocitos, otras plaquetas, las células endoteliales, las células dendríticas, los fibroblastos y las células de músculo liso vascular, entre otras. No se conoce bien el papel de esta interacción CD40L-CD40, pero sí se sabe que la interacción del CD40L de la plaqueta con el CD40 de las células endoteliales estimula la expresión y la liberación de moléculas asociadas al proceso inflamatorio15. Además, la interacción del CD40L expresado en las plaquetas con las células endoteliales de origen coronario reduce la capacidad de estas de liberar óxido nítrico (NO) y aumenta el estrés oxidativo16.
Txa2, Amplificador De La Activación PlaquetariaDespués de la activación inicial de las plaquetas, diferentes mecanismos cooperan para que esta activación se transmita al mayor número de plaquetas, y se produce lo que se conoce como fenómeno de reclutamiento plaquetario. Uno de estos factores cooperadores principales es el TxA2, que se sintetiza en la plaqueta como consecuencia de la liberación de ácido araquidónico por la acción de la fosfolipasa A217. El ácido araquidónico es el sustrato de la ciclooxigenasa-1 (COX-1). La COX-1 producirá endoperóxidos cíclicos de las prostaglandinas, PGG2 y PGH2 como productos iniciales, que se trasformarán en TxA2 por la actividad de la TxA2 sintasa. El TxA2, además de activar más plaquetas, contraerá las células del músculo liso vascular.
Es conocido que la inhibición de la COX-1 plaquetaria es el mecanismo principal de acción antiplaquetaria del ácido acetilsalicílico. El ácido acetilsalicílico acetila de forma irreversible la molécula de hidróxido (OH) de la serina en posición 529 de la COX-1, con lo que se inhibe la actividad de esta enzima. El resultado de que se reponga la actividad de COX-1 en las plaquetas depende de la producción de más plaquetas debido al carácter anucleado de estas, lo que las hace incapaces de generar nueva COX-1. Se calcula que se genera nuevo cada día aproximadamente un 10% del total de las plaquetas circulantes y que casi el 30% de las plaquetas tendrán activa la COX-1 y una producción normal de TxA2 en las 48h tras la última dosis de ácido acetilsalicílico18.
Probablemente, desde el punto de vista del tratamiento antiplaquetario, la inhibición de los receptores de TxA2 o la actividad de la TxA2 sintasa en la plaqueta tengan inicialmente una ventaja mayor que la actividad COX-1, ya que el ácido acetilsalicílico también inhibe la COX-1 endotelial que, a diferencia de la plaquetaria, libera prostaciclina, un inhibidor de la activación de las plaquetas. Se han hecho esfuerzos para desarrollar inhibidores específicos de la TxA2 sintasa; sin embargo, los resultados experimentales obtenidos han demostrado una eficiencia muy limitada en comparación con el ácido acetilsalicílico, lo que posiblemente ocurre porque al inhibirse la TxA2 sintasa se produce una acumulación de prostaciclinas G2 y H2, ambas agonistas también de los receptores de TXA2 en las plaquetas.
El ADP contribuye también a la propagación de la activación de las plaquetas. Ya hemos señalado que el ADP se libera desde los gránulos densos de las plaquetas, pero también por las células endoteliales y por los eritrocitos.
El ADP, mediante su unión a los receptores P2Y12, inhibe la adenilato ciclasa y reduce la formación de adenosinmonofosfato cíclico (AMPc) en la plaqueta, lo que facilita su activación. El ADP, por su unión a los receptores P2Y1, produce la activación de la fosfolipasa C. Estos dos receptores están acoplados a proteínas Gq y Gi respectivamente, lo que tiene gran importancia en el efecto del ADP en las plaquetas. Por ejemplo, el receptor Gq es el responsable del cambio de forma de la plaqueta inducido por ADP dependiente del reordenamiento de los filamentos de actina en la plaqueta acoplados a la activación de los receptores P2Y119.
El TxA2 también usa como mecanismo de inhibición de la activación plaquetaria la reducción en la formación de AMPc. Sin embargo, el receptor plaquetario del TxA2 no está directamente acoplado a la
proteína Gi, lo que hace que el TxA2 requiera la liberación de ADP para inhibir la actividad de adenilato ciclasa20.Implicación de la glucoproteína IIb/IIIa en la activación de las plaquetas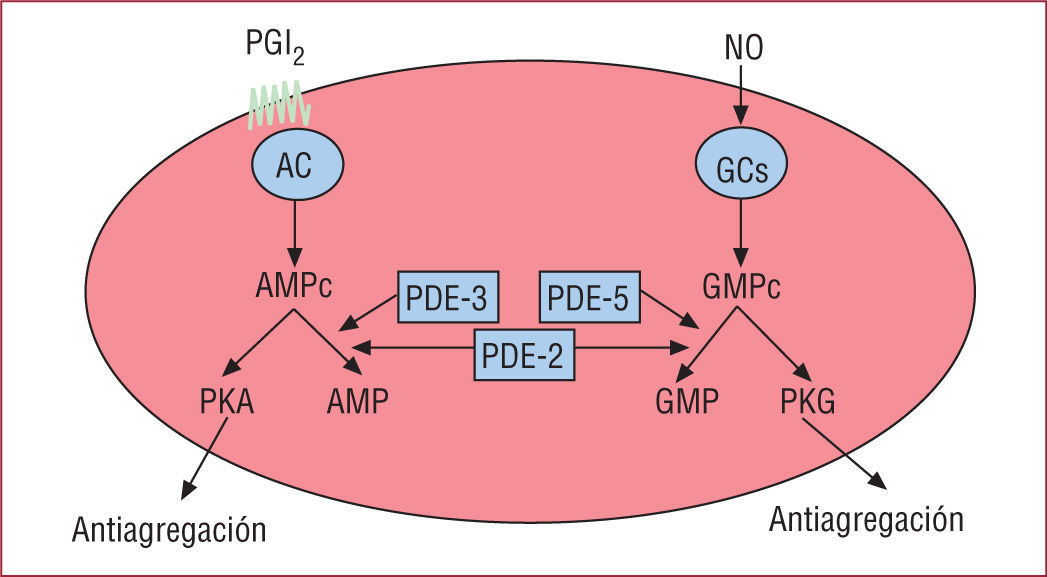
El receptor más abundante en la plaqueta es la integrina αIIbβ3 (GPIIb/IIIa) con aproximadamente 50.000–80.000 unidades en la superficie de cada plaqueta. La unión entre las subunidades GPIIb y GPIIIa ocurre mediante unión no covalente, lo que genera el receptor heterodimérico21. La mayoría de las GPIIb/IIIa se encuentran en la superficie plaquetaria, y sólo una pequeña parte se almacena en los gránulos α y en el sistema canalicular de la plaqueta, sistema de canales ramificados que se conectan a la membrana externa de la plaqueta. En las plaquetas circulantes, la GPIIb/IIIa se encuentra en estado de baja afinidad, que se transforma en alta afinidad después de la activación de estas células.
La GPIIb/IIIa interviene en la adhesión de la plaqueta a la pared vascular y también en la interacción entre plaquetas, lo que se conoce como agregación plaquetaria, mediante la interacción de dos GPIIb/ IIIa localizadas en plaquetas diferentes que se unirán entre sí a través del fibrinógeno, que hará de nexo. Aunque el fibrinógeno es el ligando principal de la GPIIb/IIIa, otras moléculas como el FvW, la fibronectina y la vitronectina se unen también a la GPIIb/IIIa21. Todas lo hacen a través de una región específica localizada en estas moléculas, conocida como región RGD por ser la nomenclatura de los aminoácidos que conforman esta región (arginina, glicina, aspártico)22. Por lo tanto, los péptidos que contienen la secuencia RGD son capaces de interaccionar con la GPIIb/IIIa e inhibir su interacción con otras moléculas. Existen otras secuencias peptídicas reconocidas por la GPIIb/IIIa, como por ejemplo lisina-glutamina-alanina-glicina-aspártico-valina. A diferencia de la región RGD, esta secuencia de 6 aminoácidos sólo se ha localizado en el fibrinógeno.
Receptores De La TrombinaLa trombina es el agonista plaquetario más potente que también facilita la producción de fibrina desde el fibrinógeno. Los receptores activados por la proteasa (PAR) son receptores de la trombina. El mecanismo de activación de los PAR y las señales que estimula son complejos. La trombina es una enzima, por lo que puede activar más de una molécula del receptor. Al unirse la trombina al receptor, ocurre una liberación proteolítica en la molécula del receptor, lo que produce la activación de cuatro tipos diferentes de proteínas G que estimulan señales diferentes en la célula23.
Hasta el momento se han descrito cuatro tipos diferentes de PAR. De ellos, solamente el PAR-1 y el PAR-4 se expresan en las plaquetas humanas y cualquiera de los dos estimula la agregación y la secreción plaquetarias24,25. El PAR-1 es el receptor principal de trombina en las plaquetas y produce la activación de la plaqueta con concentraciones bajas de trombina (EC50, 50pM), mientras que PAR-4 necesita concentraciones de trombina mayores (EC50, 5.000pM). Algunos investigadores han propuesto que en las plaquetas el PAR-1 actúa como cofactor de la activación por trombina de PAR-4, por lo que el PAR-1 y el PAR-4 actuarían como un único complejo26. Sin embargo, los estudios preclínicos indican que la inhibición del receptor PAR-1 es suficiente para la prevención del evento trombótico27. No obstante, los PAR no son de expresión exclusivamente plaquetaria, lo que dificulta el uso de este tipo de antitrombótico.
Mecanismos Endógenos De Inhibición De Las PlaquetasExisten diferentes mecanismos endógenos que pueden contrarrestar el efecto de los agonistas que inducen la activación de las plaquetas. Entre ellos, es muy probable que el NO sea el principal regulador de la activación de las plaquetas.
El NO producido por la propia plaqueta interviene en el proceso de inhibición de la agregación plaquetaria y en la reducción del reclutamiento de nuevas plaquetas al trombo formado28. En este sentido, las plaquetas de pacientes con angina inestable o infarto de miocardio producen menos NO que las plaquetas de los pacientes con angina estable, además de mostrar menos sensibilidad al NO29. Además, el ambiente prooxidante que se produce durante la fase aguda del síndrome coronario favorece una reducción en la biodisponibilidad del NO que generan otras células del entorno plaquetario, lo que reduce su efecto inhibidor en las plaquetas30.
Son muchos los mecanismos por los que el NO puede inhibir las plaquetas. El segundo mensajero principal de las acciones del NO es el GMP cíclico (GMPc). El GMPc previene la activación de las plaquetas a través de al menos tres mecanismos: a) aumenta indirectamente la concentración de AMPc por inhibición de la fosfodiesterasa 3 (PDE-3); el aumento de AMPc actúa sinérgicamente con el de GMPc para inhibir la agregación plaquetaria; b) el GMPc inhibe la activación de la fosfatidilinositol-3 cinasa (PI3K) que produce la activación de la GPIIb/ IIIa31, y c) el GMPc produce la fosforilación del receptor del TxA2 e inhibe su función.
Además, e independientemente de la producción de GMPc, el NO inhibe la exocitosis de los gránulos densos, los lisosomas y los gránulos plaquetarios32.
Se sabe que las plaquetas no activas generan cantidades de NO significativamente menores que las plaquetas activadas. En este punto hay que señalar que es muy probable que el NO liberado por la propia plaqueta no sea el principal NO que contribuya a la regulación de la actividad de estas células. Tanto el endotelio como los leucocitos circulantes, células que forman parte importante en el trombo plaquetario, también generan NO. Incluso los fármacos antiplaquetarios utilizan el NO de estas otras células para inhibir las plaquetas33,34.
La prostaciclina es otra de las moléculas endógenas que tiene una implicación directa en la regulación de la actividad plaquetaria. El producto principal derivado del ácido araquidónico en las células endoteliales es la prostaciclina producida a través del sistema de la COX y la prostaciclina sintasa. Aunque la prostaciclina tiene un efecto vasodilatador, parece que su función fisiológica principal es reducir la activación y la agregación de las plaquetas manteniéndolas en un estado no activo. En la pared vascular, la producción mayor de prostaciclina ocurre en la superficie de la íntima y disminuye según se avanza hacia la adventicia.
En la plaqueta, la activación del receptor de prostaciclina produce la estimulación de la adenilato ciclasa plaquetaria aumentando la concentración de AMPc en la plaqueta. El aumento de AMPc permite la fosforilación de la fosfoproteína estimulada por vasodilatador (VASP) y la inhibición de la movilización de calcio y la desgranulación plaquetaria. La fosforilación de VASP se ha correlacionado con la inhibición de la GPIIb/IIIa35. En este sentido, las plaquetas deficientes en VASP tienen una mayor unión del fibrinógeno a la GPIIb/IIIa36.
Hiperactividad plaquetaria y marcadores plaquetarios De actividadNo todos los pacientes tienen un estado trombótico «basal» similar. Por ejemplo, los pacientes obesos, con síndrome metabólico o diabéticos se caracterizan por tener un estado protrombótico que puede estar motivado por un aumento en la generación de trombina, una reducción de la fibrinolisis o hiperactividad plaquetaria. Se cree que este aumento de la reactividad plaquetaria basal aumenta el riesgo de aterotrombosis. Entre las alteraciones asociadas a la hiperactividad plaquetaria observada en este tipo de pacientes, se incluye mayor sensibilidad a los agonistas que inducen adhesividad y activación plaquetaria y resistencia a la respuesta antiagregante al NO37,38. Esta respuesta reducida al NO también se produce con los dadores de NO38.
Se ha considerado el tamaño del volumen plaquetario como un predictor independiente del evento vascular39. Sin embargo, no se conoce bien los mecanismos que favorecen la presencia de plaquetas más grandes. Algunos investigadores han apuntado que la existencia de un número elevado de plaquetas de mayor tamaño refleja la existencia de plaquetas reticuladas. Las plaquetas reticuladas son plaquetas jóvenes, con mayor contenido de ARN mensajero, mayor número de gránulos densos y un potencial proagregante y procoagulante aumentado40. El número de plaquetas reticuladas parece estar elevado en pacientes con síndrome coronario agudo41. Se ha propuesto la presencia de plaquetas reticuladas como un marcador de mayor recambio plaquetario. Este mayor recambio plaquetario reduce la eficacia de algunos fármacos antitrombóticos de inhibir la actividad plaquetaria42,43.
Como ya se ha referido, la plaqueta no es la única célula involucrada en el proceso trombótico. Tanto las células endoteliales como los leucocitos y los eritrocitos participan en el proceso de formación del trombo. Otras estructuras que contienen diverso material celular proveniente de estas mismas células parecen tener una importante implicación en el proceso de la activación de la plaqueta. En particular, nos referimos a las micropartículas.
Diferentes células, entre ellas las plaquetas, liberan fragmentos de 0,1–1μm, llamados micropartículas, cuya envoltura es una porción de la propia membrana plasmática celular. Las micropartículas derivadas de las plaquetas constituyen el 70–90% de las micropartículas circulantes y el aumento de su número se ha correlacionado con mayor riesgo de trombosis arterial44.
El origen celular de las micropartículas se puede identificar por la expresión de diferentes marcadores en su superficie. En concreto, para las micropartículas de origen plaquetario se ha definido la presencia del antígeno CD41+. Las micropartículas se forman tras la activación de las plaquetas por agonistas plaquetarios como la trombina y el colágeno. Sin embargo, las micropartículas positivas a marcadores que hacen sospechar su origen plaquetario también se encuentran en número elevado en sujetos inicialmente sanos cuyas plaquetas no están activadas. Además, diversos estudios que evalúan la vida media de las micropartículas han demostrado que se aclaran rápidamente de la circulación. Una posibilidad es que las micropartículas derivadas de plaquetas que se encuentran en los sujetos sanos sean consecuencia de la formación de plaquetas desde los megacariocitos.
La importancia fisiopatológica que se atribuye actualmente a las micropartículas derivadas de plaquetas es su alta capacidad protrombótica e incluso inflamatoria. La elevada actividad protrombótica de las micropartículas derivadas de plaquetas se ha asociado inicialmente con su alto contenido en fosfatidilserina, aunque el mecanismo de su trombogenicidad no está bien establecido.
Otra diana terapéutica que quizá no se ha considerado suficientemente en el área de la prevención de la trombosis arterial son los megacariocitos, células que dan origen a las plaquetas. En un artículo recientemente publicado por nuestro grupo, se observó que durante la fase aguda de un síndrome coronario, hay plaquetas cuyas propiedades proteicas hacen sospechar que se las pueda considerar «aturdidas » (bewildered)46. Estas plaquetas aturdidas existentes en esa fase aguda tienen una maquinaria asociada al metabolismo energético plaquetario reducida y una expresión menor de algunas proteínas asociadas con el citoesqueleto46. El papel fisiológico de estas plaquetas aturdidas no se conoce todavía, pero podría ser un limitador de la activación plaquetaria tras la rotura de la placa de ateroma.
Como las plaquetas tienen una vida media de aproximadamente 10 días, es difícil pensar que estas plaquetas aturdidas se hayan generado de forma aguda en número suficiente como para poder detectarlas en la masa general de plaquetas, ya que se incluyó a los pacientes en el estudio dentro de las primeras 24h desde el inicio del primer síntoma46. Por ello, y como hipótesis, es probable que días antes de romperse la placa de ateroma se estén formando este tipo de plaquetas desde los megacariocitos, probablemente inducidos por señales que incluso podrían estar relacionadas con la inflamación de la placa que se va a romper. Además, estos hallazgos nos llevan a pensar que dogmas que en principio pensamos bien establecidos, como puede ser el proceso de activación plaquetaria tras la rotura de la placa ateromatosa, pueden verse modificados al utilizar tecnologías más novedosas que aparecen constantemente y nos permiten investigar con mayor detalle los procesos fisiopatológicos.
Numerosos estudios han demostrado el efecto beneficioso del ácido acetilsalicílico en diferentes situaciones clínicas agudas, como es el caso del síndrome coronario agudo y la revascularización percutánea. Además, está suficientemente demostrado el efecto beneficioso del ácido acetilsalicílico en prevención secundaria y primaria en pacientes con factores de riesgo cardiovascular. También se ha demostrado la eficacia de la inhibición de los receptores de ADP, P2Y12, en pacientes de riesgo alto en terapia simple o en combinación con ácido acetilsalicílico. Actualmente se usan tres tienopiridinas diferentes para este propósito: la ticlopidina, el clopidogrel y el prasugrel, a las que se podría añadir el ticagrelor.
Aunque el ácido acetilsalicílico como monoterapia tiene un coste bajo y un beneficio alto, un número considerable de pacientes en tratamiento continúan sufriendo complicaciones aterotrombóticas, sobre todo los de riesgo cardiovascular elevado. También la potencia limitada del clopidogrel en su dosificación convencional ha estimulado la realización de estudios que evalúan dosis mayores. Uno de los más recientes es el estudio CURRENT-OASIS 7, que evaluó la eficacia y la seguridad de una dosis doble de clopidogrel comparada con la dosis estándar en pacientes con síndrome coronario agudo47. No se observaron diferencias significativas entre los dos regímenes terapéuticos en el objetivo primario (muerte cardiovascular, infarto de miocardio o ictus) en los 30 días de tratamiento47. Otros inhibidores de ADP, como el prasugrel, han demostrado un beneficio mayor en pacientes con diabetes mellitus y pacientes con elevación de ST sometidos a angioplastia primaria48,49. Finalmente, en el estudio PLATO se evaluó el ticagrelor, un antiagregante reversible de los receptores P2Y12 que actúa más rápidamente que el clopidogrel. En este estudio, que aleatorizó a 18.624 pacientes con síndrome coronario agudo a tratamiento con ticagrelor o clopidogrel, se observó una reducción significativa en el objetivo primario (muerte de causa vascular, infarto de miocardio o ictus) después de 12 meses de tratamiento en el brazo de tratamiento con ticagrelor50. No obstante, en este y otros estudios con otros inhibidores de los receptores de ADP como el cangrelor (CHAMPION-PCI51, CHAMPION-PLATFORM52) o el elinogrel (INNOVATE-PCI53), en los que se intentó demostrar un mayor beneficio clínico de estos con respecto al clopidogrel, es incuestionable que podemos extraer entre sus conclusiones que siguen produciéndose eventos aterotrombóticos en pacientes tratados con diferentes fármacos antiplaquetarios. Es importante, por lo tanto, profundizar en el conocimiento de los mecanismos moleculares y celulares involucrados en el proceso de activación plaquetaria, lo que nos permitirá desarrollar nuevos fármacos dirigidos a dianas terapéuticas nuevas, probablemente todavía no completamente delimitadas.
Conflicto De InteresesNinguno.