
Palabras clave
INTRODUCCIÓN
La hipertrofia ventricular izquierda (HVI), usualmente causada por hipertensión arterial, aumenta de manera considerable el riesgo de infarto del miocardio, muerte súbita, accidentes cerebrovasculares e insuficiencia cardíaca1. Modificaciones en las concentraciones de angiotensina II, catecolaminas, en algunos factores de crecimiento celular (p. ej., el factor de crecimiento análogo de insulina), endotelina y en la presión arterial (PA) contribuyen directamente al desarrollo de HVI patológica. Es posible que el desarrollo y las características moleculares de la HVI estén modulados mayoritariamente por el sistema renina-angiotensina (SRA) que por la PA propiamente.
El bloqueo farmacológico del SRA en ratas con pinzamiento (banding) de la aorta ascendente (donde la sobrecarga de presión se mantiene pero el SRA es bloqueado) disminuye en mayor grado el desarrollo de HVI que el tratamiento con el fármaco antihipertensivo hidralazina (el cual normaliza la presión arterial pero no inhibe al SRA)2.
Tanto la actividad de la enzima conversiva de la angiotensina I (ECA, dipeptidil carboxipeptidasa I, cininasa II; EC 3.4.15.1) y su ARN mensajero están incrementados en el ventrículo hipertrófico, y el aumento de la velocidad de producción de la angiotensina II se asocia con alteraciones en las propiedades diastólicas del corazón hipertrofiado3. En la hipertensión arterial, mientras el corazón desarrolla HVI patológica, podría ocurrir que la actividad de ECA circulante y/o local (tisular) aumentara. La angiotensina II y sus metabolitos son detectados en el plasma durante la administración crónica de inhibidores de ECA, lo que indica que la inhibición del SRA no es completa; esto puede limitar la eficiencia clínica de estos inhibidores en el tratamiento de la hipertensión arterial y daño cardíaco4. Existen pocos datos sobre la relación entre la actividad de ECA y la prevención precoz de la HVI hipertensiva con el uso de inhibidores de la ECA (IECA). Una mejor comprensión de esta relación ayudaría a evaluar las ventajas terapéuticas de la inhibición exlusiva de la ECA o del bloqueo del SRA en otro nivel (p. ej., utilizando bloquedores del receptor tipo 1 de la angiotensina II o bloquedores del receptor de aldosterona) en la prevención y tratamiento de esta condición patológica. No existen estudios del efecto de los IECA en la prevención precoz de la HVI inducida por sobrecarga de presión ni tampoco una evaluación simultánea de las concentraciones de angiotensina II. Hemos evaluado aquí el efecto del IECA perindopril administrado de forma precoz y a dosis conocidamente antihipertensivas y capaces de inhibir la ECA sobre los valores de presión arterial, HVI, actividad de la ECA circulante y también sobre las concentraciones de angiotensina II en el plasma y el ventrículo izquierdo en el modelo Goldblatt en la rata (2 riñones-un pinzado, 2r-1p) a las 2 semanas de la intervención quirúrgica.
MATERIAL Y MÉTODO
Modelo experimental de hipertrofia ventricular izquierda hipertensiva
Para los experimentos se siguieron las recomendaciones de la Guía para el Cuidado y Uso de Animales de Laboratorio (publicación NIH Nº 85-23, revisada en 1985) y aprobada por la Comisión de Investigación de la P. Universidad Católica de Chile. La hipertensión experimental y la HVI fueron inducidas en la rata usando el modelo Goldblatt (Gb; 2r-1p) como se ha descrito con anterioridad5. Brevemente, bajo anestesia con éter, ratas macho Sprague-Dawley (80-100 g) fueron sometidas a una pequeña incisión abdominal tras la cual se colocó una pinza de plata (0,2 mm de espesor interno) alderedor de la arteria renal izquierda, dejando la arteria renal derecha intacta. Todas las ratas sobrevivieron a la operación e ingresaron en el estudio. Los animales fueron mantenidos bajo condiciones controladas de luz y oscuridad y tuvieron libre acceso a agua y comida. Las ratas del grupo control fueron sometidas a la misma incisión abdominal pero sin la colocación de la pinza alrededor de la arteria renal izquierda.
La presión arterial sistólica (PAS) se midió con método pletismográfico en la cola (tail cuff) 24 h antes del sacrificio. Las ratas fueron pesadas y decapitadas. Los corazones fueron rápidamente extraídos. La aorta, la arteria pulmonar y los ventrículos fueron separados con cuidado y lavados extensamente con solución salina fría para limpiar toda la sangre. El grado de HVI se cuantificó usando la relación entre el peso del ventrículo izquierdo y la masa corporal (masa ventricular izquierda relativa [MRVI VI] * 100/peso corporal).
Grupos experimentales
Se usaron 3 grupos experimentales: Gb, control y Gb-Per (11 animales por grupo). El IECA perindopril (Per) se administró en dosis de 4 mg/kg/día por gavaje diario, comenzando inmediatamente después de la cirugía y continuando durante 2 semanas. El día del sacrificio se recolectó sangre para medir la actividad de la ECA plasmática y las concentraciones de angiotensina II. Los ventrículos izquierdos se lavaron exhaustivamente en una solución salina, se congelaron en nitrógeno líquido y se almacenaron a -80 ºC para la determinación de la angiotensina II tisular.
Determinación de la actividad de la ECA en el plasma y el ventrículo izquierdo
La acividad de la ECA se midió por fluorimetría por medio de la hidrólisis de Z-fenil-L-histidil-L-leucina (Bachem Bioscience Inc., EE.UU.), un sustrato análogo de la ECA6-8. El producto L-histidil-L-leucina se cuantificó por espectrofluorimetría, para lo cual se usó un espectrofotómetro luminiscente (Perkin Elmer LS 50b, EE.UU.). Las determinaciones se realizaron por duplicado. La actividad de la ECA plasmática se expresó en U/ml (1 U = 1 nmol L-histidil-L-leucina/min). Para la medición de la actividad de la ECA tisular, 200 mg del ventrículo izquierdo se homogeneizaron con tampón tris-HCl, 50 mmol/l; pH 8,0; NaCl 1%. El extracto se centrifugó a 1.000 g por 15 min a 4 ºC y el sobrenadante resultante se recentrifugó a 105.000 g por 30 min a 4 ºC. La pella se resuspendió en tampón CHAPS 8 mmol/l. La concentración de proteínas se midió por Bradford usando seroalbúmina de bovino como estándar9. La actividad de ECA tisular se expresó en U/mg de proteína.
Recolección de muestras para la medición de la angiotensina II plasmática
Las muestras de sangre para la medición de la angiotensina II se recogieron en tubos de poliestireno preenfriados, como describen Admiraal et al10, fueron inmediatamente transferidas a tubos de poliestireno y centrifugadas a 3.000 g por 10 min a 4 ºC. Las muestras de plasma se almacenaron a -80 ºC y se determinaron dentro de la semana siguiente. Los plasmas fueron directamente aplicados en cartridges Sep-pack, como se describe a continuación.
Recolección, extracción y determinación de la angiotensina II en el ventrículo izquierdo
Los corazones se extrajeron rápidamente y las aurículas y los ventrículos derechos e izquierdos fueron separados. Estos tejidos se congelaron inmediatamente en nitrógeno líquido y se guardaron a -80 ºC hasta su determinación. Los ventrículos izquierdos se homogeneizaron por Ultraturrax en solución de extracción fría. Los homogeneizados se centrifugaron a 4 ºC y los sobrenadantes se concentraron por evaporación. El remanente de cada sobrenadante se diluyó en 0,1% H3PO4 y se recentrifugó a 4 ºC. El sobrenadante se diluyó con 0,02% H3PO4 y se concentró por adsorción reversible en cartridges de octadecil silil sílica (SepPak C-18, Waters, Milford, MA). Los cartridges se activaron con metanol y se equilibraron con 2 lavados de agua destilada fría. Las muestras se aplicaron a los cartridges a 4 ºC, seguido de 2 lavados con agua destilada fría y con hexano (sólo para las muestras de tejido). Las angiotensinas adsortas se eluyeron con metanol al 90% y, posteriormente, el metanol se evaporó a 4 ºC, usando un concentrador Speed-Vac. Las angiotensinas se separaron por HPLC de fase reversa con el método de Nussberger et al11, usando una columna Bondapak C18. El extracto concentrado del SepPak se disolvió antes de la inyección. Los eluidos se recogieron en tubos de poliestireno en fracciones que contenían BSA al 0,1%. Las fracciones que contenían angiotensina II se neutralizaron con 0,5 mol/l de NaOH. Las concentraciones de angiotensina II se midieron por radioinmunoanálisis12. La recuperación de la angiotensina II después de la homogeneización, extracción y separación por HPLC se determinó agregando [125I]angiotensina II al tejido (10.000 cpm/g) antes de la homogeneización.
Análisis estadístico
Los resultados se expresan como media ± error estándar. Para las comparaciones múltiples se usó el análisis de la variancia con un factor (ANOVA) y posteriormente la prueba de Student-Newman-Keuls. También se utilizó el test de correlación lineal. Un valor de p <= 0,05 fue considerado como estadísticamente significativo.
RESULTADOS
Estudios hemodinámicos e hipertrofia ventricular izquierda
Como se expone en la tabla 1, se observó hipertensión arterial a las 2 semanas en el grupo Gb (F = 54; p < 0,001) comparado con el grupo control. También se observó un grado significativo de HVI (20% de aumento en MRVI comparado con las ratas controles) en el grupo Gb (F = 11,6; p < 0,001). Ambos efectos fueron totalmente prevenidos por la administración de perindopril (tabla 1). Se observaron pequeñas diferencias en la masa corporal en los animales controles comparados con las ratas Gb. Se observó una correlación significativa entre la PAS y la MRVI (r = 0,51; p < 0,01).

Actividad de la ECA plasmática y tisular
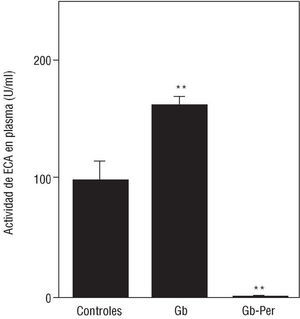
Las actividades de ECA en plasma, pulmón y ventrículo izquierdo a las 2 semanas de la intervención quirúrgica se exponen en la figura 1. La actividad de la ECA plasmática fue un 60% mayor en las ratas Gb comparada con sus respectivos controles (fig. 1A) y en el ventrículo izquierdo fue 3 veces mayor en el grupo Gb que en los animales del grupo control (p < 0,001) (fig. 1B). No hubo diferencias en las actividades de la ECA pulmonar entre los grupos Gb y control (fig. 1C).

Fig. 1. Actividad de la enzima conversiva de la angiotensina (ECA) en plasma (A), ventrículo izquierdo (B) y pulmón (C) durante el desarrollo precoz de hipertrofia ventricular izquierda (HVI) hipertensiva. La actividad de ECA está expresada en U/ml de plasma o U/mg de proteína tisular. 1 U: 1 nmol L-histidil-L-leucina/min. Controles (n = 11) y Goldblatt (Gb; n = 11). Valores expresados como media ± error estándar. *p < 0,05 o **p < 0,01 frente a grupo control (post-ANOVA).
No se encontró correlación entre la PAS y la actividad de la ECA en el plasma o el ventrículo izquierdo. La MRVI no se correlacionó con la actividad de la ECA plasmática.
Concentraciones de angiotensina II en el plasma y el ventrículo izquierdo
En experimentos preliminares con 2 inhibidores diferentes de la ECA no encontramos inhibición de la actividad de la ECA en el ventrículo izquierdo, mientras que simultáneamente observamos una reducción del 99% en la actividad de la ECA plasmática (datos no expuestos). Por esta razón, hemos medido las concentraciones de angiotensina II en el plasma y el ventrículo izquierdo después de 2 semanas de tratamiento con perindopril (4 mg/kg/día, vía gavaje) para estimar así el grado de inhibición de la ECA en el ventrículo izquierdo. Campbell et al encontraron que la dosis de perindopril utilizada en el presente estudio inhibe casi completamente la actividad de la ECA plasmática12, lo cual hemos confirmado en experimentos preliminares.
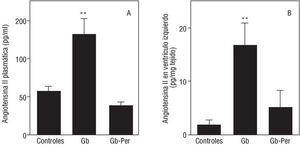
Las concentraciones de angiotensina II en el plasma y el ventrículo izquierdo (fig. 2) aumentaron de manera significativa en las ratas Gb a las 2 semanas de la intervención quirúrgica (129 y 800%, respectivamente; F = 12,2; p < 0,01 y 5,9; p < 0,01) y también se corre lacionaron significativamente entre sí (r = 0,6; p < 0,002). La actividad de la ECA circulante se correlacionó de manera significativa con las concentraciones plasmáticas de angiotensina II (r = 0,51; p < 0,02).

Fig. 2. Efecto de la administración precoz del inhibidor de la enzima conversiva de la angiotensina (ECA) perindopril sobre la actividad de la ECA plasmática durante la prevención precoz de hipertrofia ventricular izquierda (HVI) hipertensiva. Se determinó la actividad de la ECA plasmática (U/ml) en los grupos control (n = 11), en las ratas Goldblatt a las 2 semanas de la intervención (Gb, n = 11) y en las ratas tratadas con perindopril (4 mg/kg/día, administrado por gavaje diario) (Gb-Per, n = 11). Valores expresados como media ± error estándar. **p < 0,05 frente a grupo control (post-ANOVA). Gb: Goldblatt, Per: perindopril.
La administración preventiva de perindopril durante 2 semanas comenzando inmediatamente después de la cirugía inhibió completamente la actividad de la ECA circulante (fig. 3), causando una gran disminución de las concentraciones de angiotensina II en el plasma y en el ventrículo izquierdo (fig. 2). Las concentraciones de angiotensina II plasmática se correlacionaron significativamente con la PAS (r = 0,52; p < 0,005) y con la MRVI (r = 0,48; p < 0,005). Los valores de angiotensina II en el ventrículo izquierdo se correlacionaron con la PAS (r = 0,5; p < 0,01) y con la masa ventricular izquierda (r = 0,41; p < 0,03).

Fig. 3. Efecto de la administración precoz del IECA perindopril sobre las concentraciones de angiotensina II en el plasma y el ventrículo izquierdo durante la prevención precoz de la hipertrofia ventricular izquierda (HVI) hipertensiva. Las concentraciones de angiotensina II en el plasma y el ventrículo izquierdo (pg/ml y pg/mg tejido, respectivamente) se midieron después de la administración de perindopril (4 mg/kg/día, administrado por gavaje diario), que se inició inmediatamente después de la cirugía y se continuó durante 2 semanas (Gb-Per). Las concentraciones de angiotensina II en el plasma (A) y en el ventrículo izquierdo (B) se midieron por HPLC y radioinmunoanálisis (véase el apartado Material y métodos). Valores expresados como media ± error estándar. **p < 0,05 fr ente a grupo control (post-ANOVA). Gb: Goldblatt; Per: perindopril.
DISCUSIÓN
Estos resultados ponen de manifiesto que: a) la actividad de la ECA y las concentraciones de angiotensina II en el plasma y el ventrículo izquierdo aumentan precozmente en la HVI hipertensiva en este modelo experimental, y b) el uso preventivo de un inhibidor de la ECA (en este caso perindopril) normalizó la actividad de la ECA y las concentraciones de angiotensina II en el plasma y en el ventrículo izquierdo, y además previno el desarrollo de HVI, lo que se asoció a una normalización de la presión arterial.
La ECA es una zinc metalopeptidasa unida a membrana que está localizada principalmente en el endotelio de los vasos, sobre todo en la circulación pulmonar13. También se encuentra en la superficie de algunas células epiteliales, en las células mononucleares circulantes, macrófagos, células germinales masculinas y en el plasma o suero13. La ECA circulante proviene probablemente de células vasculares endoteliales. En el plasma y/o en células endoteliales la ECA convierte el decapéptido inactivo angiotensina I en el octapéptido activo angiotensina II13. En humanos, las concentraciones de ECA en el plasma varían durante el crecimiento: son elevadas durante la infancia y luego disminuyen, para permanecer estables durante la etapa adulta14.
En la hipertensión arterial, el aumento de las concentraciones de angiotensina II induce fibrosis miocárdica e HVI patológica15-17. Incluso a concentraciones subhipertensivas, la angiotensina II induce daño y fibrosis miocárdica18. La angiotensina II es también un potente vasoconstrictor que estimula la síntesis de proteínas en el corazón aislado de rata19, la secreción de aldosterona y el crecimiento tanto del músculo liso como de miocitos cardíacos. La angiotensina II también estimula la síntesis de colágeno tipo I en células intersticiales en cultivo20. Además de la angiotensina II existen otros factores que estimulan la fibrogénesis en el miocardio, como la aldosterona, la endotelina y el factor de transformación tisular beta. Por otro lado, existen factores que contrarrestan los factores mencionados previniendo el desarrollo de fibrosis en el miocardio, como la bradicinina, el óxido nítrico, el péptido natriurético auricular y los glucocorticoides21. Es posible que genotipos que determinen concentraciones más elevadas de factores estimuladores (o menores valores de factores inhibidores) de fibrosis miocárdica puedan producirla con mayor facilidad frente a un mismo estímulo fibrogénico.
Challah et al observaron un aumento de la actividad de la ECA plasmática después de 2 semanas de la cirugía (pero no después de 4 y 12 semanas) usando el mismo modelo experimental que nosotros22. En la HVI hipertensiva establecida (Gb 9 semanas) nosotros observamos previamente que hay una activación de la ECA en el ventrículo izquierdo pero no en el plasma (datos no expuestos). Un aumento en la actividad de la ECA plasmática indica un incremento en la forma secretada de la ECA (ECA soluble), pero el origen de este aumento endotelial o de otras vías aún no ha sido determinado completamente.
Un aumento de la actividad de la ECA en el ventrículo izquierdo y de la expresión de su correspondiente ARN mensajero también se ha encontrado después de 2 y 4 semanas de cirugía, pero no después de 12 semanas en el mismo modelo experimental22. Varios estudios han demostrado que la actividad de la ECA en el ventrículo izquierdo aumentó en diferentes modelos de hipertrofia patológica23-25. Por ejemplo, en ratas con infarto al miocardio experimental, un aumento de la actividad de la ECA en el septum interventricular se debe a la inducción de su ARN mensajero y a una mayor síntesis local26. En nuestro estudio, el uso de un inhibidor de la ECA (perindopril) por un corto período de tiempo previno el desarrollo de HVI, lo que se asoció a una disminución de la presión arterial y a una inhibición simultánea de la actividad de la ECA circulante y en el ventrículo izquierdo.
Uno de los aspectos morfológicos característicos de la HVI patológica es el aumento del colágeno perivascular e intersticial27,28 con disfunción diastólica temprana y sistólica tardía del ventrículo izquierdo29-33. En la HVI hipertensiva, la sobreproducción de ECA en el miocardio parece indicar un remodelamiento intersticial, pero no hipertrofia de los cardiomiocitos22. Las células miocárdicas intersticiales responsables del aumento de la actividad de la ECA podrían ser activadas por fibroblastos, células inflamatorias, células musculares lisas activadas o todas ellas. Por tanto, en nuestro grupo Gb no tratado la activación por algunas de estas células podría ser la responsable del aumento de la actividad de la ECA en el ventrículo izquierdo.
La disminución de la actividad de la ECA en el ventrículo izquierdo durante la inhibición de la ECA ha sido observada previamente en ratas normales tratadas con quinapril34, en ratas con pinzamiento de la aorta abdominal35 y en ratas espontáneamente hipertensas36. También se ha observado usando el IECA ramipril en ratas transgénicas (TGR; mREN2) con hipertensión crónica37 y, además, con enalapril y perindopril en ratas postinfarto de miocardio38,39.
Investigaciones anteriores han determinado las concentraciones de angiotensina II en el ventrículo izquierdo en ratas postinfarto de miocardio tratadas con perindopril40, también en ratas con fístula aortocava tratadas con quinapril41 pero no en la prevención precoz de HVI hipertensiva en el modelo Goldblatt. Esta observación tiene implicancias clínicas potenciales, ya que enfatiza la prevención (o el retardo de su desarrollo) de la HVI patológica en la hipertensión arterial dependiente de angiotensina II utilizando un IECA.
Resultan llamativos los cambios en la masa ventricular derecha con el tratamiento preventivo, ya que ésta disminuye significativamente con perindopril. En esta cavidad sólo deja de actuar como estímulo hipertrófico la angiotensina II, a diferencia de lo que sucede con la prevención de la HVI, en que en el ventrículo izquierdo dejan de actuar dos estímulos hipertrofiantes, como la sobrecarga hemodinámica y la angiotensina II. Al respecto, se ha sugerido que la hipertrofia miocitaria sería más dependiente de la sobrecarga hemodinámica, mientras que la angiotensina II y la aldosterona regularían la acumulación de colágeno en ambos ventrículos42.
En este estudio no hemos determinado directamente la actividad de ECA en el ventrículo izquierdo debido a que en experimentos piloto previos no pudimos detectar su inhibición, mientras que simultáneamente observamos < 1% de actividad de ECA en plasma (datos no expuestos). Esta subestimación de la inhibición de la ECA en el ventrículo izquierdo se podría explicar por el hecho de que el procedimiento puede tomar en nuestro caso 3 h a partir del momento de la remoción del tejido hasta el final de la determinación. Durante este período, los IECA unidos a la ECA tisular podrían disociarse y subestimarse así el grado verdadero de inhibición de ECA, como ha sido previamente comunicado por Urata et al43 y Kinoshita et al44. La medición de angiotensina II en el ventrículo izquierdo se ha considerado como una forma precisa para determinar la actividad de ECA al utilizar IECA en tejido cardíaco12,43,45. También es posible que el perindopril, igual que otros IECA, adicionalmente pueda actuar inhibiendo la degradación de bradicinina12. Algunos estudios, sin embargo, han demostrado que la prevención de HVI secundaria a hipertensión vasculorrenal por IECA es independiente del sistema renina-angiotensina y de la presión arterial46,47.
Limitaciones del estudio
Hubiera sido interesante incluir un grupo de ratas hipertensas tratadas con una dosis no hipotensora de perindopril para dilucidar el mecanismo de prevención de la HVI. En el modelo utilizado descienden simultáneamente la presión arterial y las concentraciones circulantes y miocárdicas de angiotensina II tras la administración de perindopril, lo cual hace más difícil delimitar la contribución de cada uno de ambos mecanismos al efecto del fármaco sobre la HVI. Ésta es una limitación del estudio, que estuvo diseñado para evaluar el efecto de dosis conocidamente antihipertensivas y capaces de inhibir la ECA de perindopril sobre la prevención precoz de la HVI.
Conclusiones
En este modelo experimental de HVI hipertensiva dependiente de angiotensina II existe una activación precoz de la ECA circulante y en el ventrículo izquierdo. La administración precoz de un IECA en una dosis antihipertensiva inhibe la ECA circulante (y muy probablemente la ECA en el ventrículo izquierdo), normaliza las concentraciones elevadas de angiotensina II en el plasma y el ventrículo izquierdo y previene (pudiendo eventualmente regresar y retardar) la aparición de HVI.
Financiado parcialmente por Fondecyt Proyectos 1961065 y 2970021, por un Programa de Intercambio ECOS-Conicyt y por una beca internacional de la Uchida Foundation. Correspondencia: Dr. J.E. Jalil. Pontificia Universidad Católica de Chile. Facultad de Medicina. Departamento de Enfermedades Cardiovasculares. Marcoleta, 367. Santiago de Chile. Recibido el 30 de octubre del 2000.
Aceptado para su publicación el 24 de mayo del 2001.