
Nuestro objetivo fue aproximar la prevalencia de mutaciones en los genes candidatos de hipercolesterolemia familiar (HF) en una población española de mediana edad, y determinar el valor predictivo de los criterios clínicos de sospecha de HF en la detección de mutaciones causales.
MétodosSe seleccionaron individuos mayores de 18 años no relacionados de la cohorte Estudio de Salud de los Trabajadores de Aragón (AWHS) con colesterol unido a lipoproteínas de baja densidad (cLDL)> percentil 95, con enfermedad cardiovascular prematura o con cLDL> 130 mg/dl con tratamiento hipolipemiante, asumiendo que al menos una de las características estará presente en todos los individuos con HF. En estos participantes se secuenciaron los genes LDLR, APOB, PCSK9, APOE, STAP1 y LDLRAP1 mediante secuenciación masiva.
ResultadosDe 5.400 individuos del AWHS, 4.514 tenían datos lipídicos y registro farmacológico hipolipemiante completo, 255 participantes (5,65%) cumplían los criterios de sospecha de HF, 24 de ellos (9,41%) fueron diagnosticados de hiperlipoproteinemia(a) y 16 (6,27% de los secuenciados) presentaron alguna mutación causal en genes candidatos: 12 participantes portaban 11 alelos patogénicos diferentes en LDLR y 4 participantes portaban una mutación patogénica en PCSK9. Las concentraciones de cLDL> 220mg/dl y el cLDL> 130mg/dl a pesar del tratamiento con estatinas mostraron la mayor asociación con la presencia de mutación (p=0,011).
ConclusionesNuestros resultados muestran que la prevalencia española de HF es 1:282 y sugieren que una concentración de cLDL elevado, y niveles altos de cLDL a pesar de la terapia con estatinas son los mejores predictores para un diagnóstico genético positivo de HF.
Palabras clave
La hipercolesterolemia familiar (HF) es un trastorno genético caracterizado por unas concentraciones plasmáticas de colesterol total muy altas, debidas al aumento del colesterol unido a las lipoproteínas de baja densidad (cLDL), con un gran riesgo de enfermedad coronaria (EC) prematura1. Tradicionalmente, la HF se ha descrito como una enfermedad monogénica con una transmisión autosómica codominante y una prevalencia estimada de alrededor de 1:500 en la población general2. Sin embargo, hay algunas discrepancias por lo que respecta a la prevalencia de la HF. Estudios recientes han puesto de manifiesto que la HF clínica probablemente sea más frecuente de lo que se había pensado, 1:217 en el estudio Copenhagen General Population3. Un diagnóstico temprano es crucial en la HF, ya que se ha demostrado que el tratamiento hipolipemiante reduce drásticamente la EC4, sobre todo si el tratamiento se inicia en una fase temprana de la vida5. No obstante, en la población general la HF está infradiagnosticada e infratratada6.
El diagnóstico clínico de la HF se basa en una alta concentración plasmática de cLDL, antecedentes familiares de hipercolesterolemia, antecedentes personales y familiares de EC prematura y signos de depósito de colesterol, como xantomas tendinosos y arco corneal prematuro. Estos parámetros se evalúan a menudo con puntuaciones clínicas aplicando los criterios Make Early Diagnosis to Prevent Early Death (MEDPED), la modificación del MEDPED de la Dutch Lipid Clinic Network (DLCN) o los criterios del Simon Broome Register Group (SBRG)7. Se ha demostrado que estos criterios clínicos muestran una intensa asociación con el diagnóstico genético8,9. Sin embargo, estos criterios incluyen una información que no siempre está disponible o requiere cierto conocimiento experto, y se emplean la mayoría de las veces en unidades especializadas6,10. Por esta razón, y con objeto de mejorar el diagnóstico, el European Atherosclerosis Society Consensus Panel recomienda unos criterios de sospecha, previos al diagnóstico clínico: a) en los adultos, un colesterol total en plasma > 310mg/dl o > percentil 95 correspondiente a la edad y el sexo en el país; b) en los niños, una concentración de colesterol total en plasma > 230 mg/dl o > percentil 95 correspondiente a la edad y al sexo en el país; c) EC prematura (a una edad < 55 años los varones y < 60 años las mujeres); d) xantomas tendinosos; e) muerte súbita por EC prematura de un familiar (a una edad < 55 años los varones y < 60 años las mujeres)6. Sin embargo, no se ha validado la utilidad clínica de estos criterios de sospecha.
El diagnóstico genético es el patrón de referencia en una enfermedad monogénica y se recomienda encarecidamente su uso en la HF11. La HF tiene origen en mutaciones de diversos genes: LDLR, que codifica para el receptor de las lipoproteínas de baja densidad (LDL); APOB, que codifica para la apolipoproteína B12, y PCSK9, para la enzima proproteína convertasa subtilisina/kexina tipo 913. Se ha identificado un nuevo locus causante de HF: la mutación p.(Leu167del) en el gen APOE14. Además, varias mutaciones en el gen STAP1, que forma parte de la familia del adaptador de transducción de señal, se han asociado con la HF15, si bien recientemente se ha puesto en duda el papel de este gen16,17. Por otra parte, las concentraciones de lipoproteína(a) [Lp(a)] en plasma son un rasgo heredable que se asocia con un aumento del riesgo de EC, y se ha descrito que la hiperlipoproteinemia(a) [hiperLpa(a)] es una causa frecuente de hipercolesterolemia autosómica dominante18. El conocimiento de la mutación causal confirma el diagnóstico, establece un pronóstico, facilita los exámenes de detección sistemática en la familia y mejora la adherencia del paciente al tratamiento19. La presencia de mutaciones patógenas en los genes causantes de la HF en pacientes con un diagnóstico clínico de HF implica un aumento sustancial del riesgo de EC20, por lo que las pruebas genéticas permiten la identificación de las personas que tienen el mayor riesgo de EC de entre las que presentan hipercolesterolemia. Sin embargo, estas pruebas genéticas tienen también ciertas dificultades, como son la complejidad genética de la enfermedad, con la implicación de varios genes, miles de mutaciones diferentes, algunas de las cuales tienen una trascendencia patógena incierta, y la poca disponibilidad del análisis genético en algunos sistemas de salud.
El objetivo del presente estudio es estimar la prevalencia de las mutaciones en genes candidatos en una población de mediana edad de España mediante criterios de sospecha clínica, y analizar su valor predictivo respecto a la presencia de mutaciones causales.
MÉTODOSParticipantesSe seleccionó a los individuos evaluados de la población del Aragon Workers’ Health Study (AWHS). El AWHS es un estudio de cohorte longitudinal de los factores de riesgo cardiovascular y de la ateroesclerosis subclínica que se lleva a cabo en 5.400 trabajadores de la fábrica Opel España, situada en Aragón (España), y que han sido objeto de seguimiento desde 200921. Para esta publicación se estudió a los individuos no emparentados de edad ≥ 18 años del AWHS de los que se dispusiera de determinaciones anuales o bianuales desde 2009. Se utilizaron los siguientes criterios para seleccionar a las personas con sospecha clínica de HF: a) participantes con concentraciones de cLDL no tratadas > percentil 95 de la población española estratificada según la edad y el sexo22 en como mínimo 2 determinaciones; b) participantes con EC prematura (edad < 55 años los varones y < 60 años las mujeres) en la situación inicial; c) participantes en tratamiento con estatinas y con cLDL > 130mg/dl en al menos 2 determinaciones, y d) participantes con concentraciones de cLDL no tratadas > percentil 95 de la población española en tan solo 1 determinación pero también el cLDL > 130mg/dl en tratamiento con estatinas. Los criterios de exclusión fueron la presencia basal o durante el seguimiento de causas secundarias de hipercolesterolemia, como obesidad grave (índice de masa corporal [IMC] > 35), diabetes mellitus tipo 2 mal controlada (HbA1c > 8%), enfermedad renal con tasa de filtrado glomerular < 30ml/min y/o macroalbuminuria, hepatopatía (alanina transaminasa > 3 veces el límite superior de la normalidad), hipotiroidismo (tirotropina > 6 mUI/l), embarazo, enfermedades autoinmunitarias y el tratamiento con inhibidores de proteasa. Se definió al sujeto con hiperLp(a) como el individuo con sospecha de HF y una concentración de Lp(a) > 50mg/dl, pero concentraciones de cLDL ajustadas por la Lp(a) normales (< percentil 95 de la población española)22.
Todos los participantes dieron su consentimiento informado por escrito y el estudio fue aprobado por el Comité de Ética de Investigación Clínica de Aragón (CEICA).
Se llevó a cabo una evaluación de los factores de riesgo cardiovascular, los antecedentes personales y familiares de enfermedad cardiovascular, la toma de medicamentos con efectos en el metabolismo lipídico y los parámetros antropométricos de todos los participantes en la situación inicial y luego a intervalos anuales o bianuales durante el seguimiento.
Análisis de lípidosSe realizaron análisis de lípidos y lipoproteínas en muestras de plasma en EDTA obtenidas tras una noche o al menos 10 h en ayunas, a intervalos anuales o bianuales desde el año 2009. Se incluyeron las determinaciones realizadas hasta 2015. Se determinaron las concentraciones de colesterol total y de triglicéridos con métodos enzimáticos estándares. El colesterol unido a lipoproteínas de alta densidad (cHDL) se determinó directamente con una reacción enzimática de la colesterol oxidasa (UniCel DxC 800; Beckman Coulter, Inc., Estados Unidos). El cLDL se calculó con la fórmula de Friedewald. Los valores lipídicos de los participantes que estaban en tratamiento hipolipemiante se ajustaron en función del tratamiento con estatinas23. Ninguno de ellos estaba een tratamiento con inhibidores de la PCSK9. La Lp(a) se determinó mediante nefelometría cinética IMMAGE (Beckman Coulter Inc., Estados Unidos). Se obtuvieron los valores de cLDL ajustados por la Lp(a) restando a la concentración de cLDL calculada con la fórmula de Friedewald la concentración de Lp(a) dividida por 324,25.
Análisis genéticoSe aisló el ADN genómico de sangre total mediante métodos estándares. Mediante secuenciación de nueva generación con la plataforma SeqProLipo Platform (Progenika Biopharma Grifols, España), de secuenciaron los genes LDLR (NM_000527.4), PCSK9 (NM_174936.3), APOE (NM_000041.3), STAP1 (NM_0121108.3) y LDLRAP1 (NM_015627.2) y los exones 26 y 29 del gen APOB (NM_000384.2), que contienen el código del lugar de unión LDLR. Dicha plataforma incluye mutaciones puntuales, reordenaciones grandes y variaciones en el número de copias.
Para evaluar la patogenicidad de las variantes identificadas, se utilizó SIFT26, PolyPhen-227, Mutation Taster28 y PredictSNP29. El efecto de las variantes en los posibles lugares de corte y empalme se predijo con FruitFly30. Para comparar las frecuencias de las variantes identificadas en los participantes en el estudio y en la población general, se compilaron las frecuencias alélicas de las variantes identificadas procedentes del 1000 Genomes Project31 y los ExAc Browser Data32. Las variantes raras se definieron como las variantes genéticas con una frecuencia <1% de la población general. Una variante rara se definió como variante causal cuando ya estaba asociada con la HF y como variante posiblemente patógena cuando la predicción del análisis bioinformático la clasificaba como patógena.
Análisis estadísticoLos análisis se realizaron con el programa informático de cálculo estadístico R versión 3.5.033. La distribución de las variables se analizó con la prueba de Shapiro. Las variables cuantitativas con una distribución normal se expresan en forma de media ± desviación estándar y se analizaron con una prueba de ANOVA. Las variables con una distribución asimétrica se expresan como mediana [intervalo intercuartílico] y se analizaron con la prueba de Kruskal-Wallis. Las variables cualitativas se expresan en forma de porcentajes y se analizaron con la prueba de la χ2. El nivel de significación estadística se estableció en p < 0,05. Se utilizó una regresión binaria logística para determinar el mejor diagnóstico con los criterios de la European Atherosclerosis Society, tomando la presencia de una mutación en genes candidatos como variable dependiente.
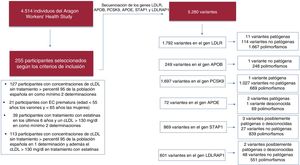
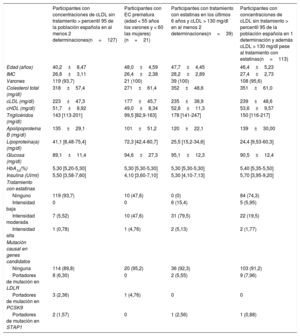
RESULTADOSParticipantes en el estudio que cumplían los criterios clínicos de HFDe los 5.400 individuos incluidos en el AWHS, se dispuso de la información necesaria para evaluar los criterios de inclusión y exclusión de 4.514. Se excluyó a un total de 886 individuos porque los datos lipídicos registrados no eran completos, tenían menos de 2 paneles lipídicos completos y/o datos incompletos sobre el uso de fármacos hipolipemiantes. De los 4.514 participantes, 255 (5,65%) cumplían los siguientes criterios de inclusión (que no son mutuamente excluyentes): a) 127 participantes presentaban concentraciones de cLDL no tratadas > percentil 95 de la población española en al menos 2 determinaciones; b) 21 tenían una EC prematura en la situación inicial (edad < 55 años los varones y < 60 las mujeres); c) 39 tenían una concentración de cLDL > 130mg/dl en como mínimo 2 determinaciones pese al tratamiento con estatinas, y d) 113 tenían una concentración de cLDL sin tratamiento > percentil 95 de la población española en 1 determinación y también un cLDL > 130mg/dl en al menos otra ocasión en tratamiento con estatinas (figura 1).

Los participantes con concentraciones de cLDL altas a pesar del tratamiento con estatinas y los que tenían una EC en la situación inicial eran de más edad y tenían un IMC más alto que el resto de los participantes. Los participantes que estaban en tratamiento con estatinas y tenían concentraciones de cLDL altas a pesar del tratamiento farmacológico y aquellos con cLDL elevado en 1 determinación y también concentraciones de cLDL altas en tratamiento con estatinas presentaban unas concentraciones de colesterol total, cLDL y triglicéridos más altas que las de los demás participantes. Los participantes con una EC prematura tenían concentraciones de Lp(a) más altas. Los participantes con concentraciones altas de cLDL en como mínimo 2 determinaciones y que no tomaban ningún tratamiento hipolipemiante fueron el 93,7% (tabla 1).
Características bioquímicas y clínicas basales y número de portadores de mutación según los criterios de selección
| Participantes con concentraciones de cLDL sin tratamiento > percentil 95 de la población española en al menos 2 determinaciones(n=127) | Participantes con EC prematura (edad < 55 años los varones y < 60 las mujeres)(n=21) | Participantes con tratamiento con estatinas en los últimos 6 años y cLDL > 130 mg/dl en al menos 2 determinaciones(n=39) | Participantes con concentraciones de cLDL sin tratamiento > percentil 95 de la población española en 1 determinación y además cLDL > 130 mg/dl pese al tratamiento con estatinas(n=113) | |
|---|---|---|---|---|
| Edad (años) | 40,2±8,47 | 48,0±4,59 | 47,7±4,45 | 46,4±5,23 |
| IMC | 26,8±3,11 | 26,4±2,38 | 28,2±2,89 | 27,4±2,73 |
| Varones | 119 (93,7) | 21 (100) | 39 (100) | 108 (95,6) |
| Colesterol total (mg/dl) | 318±57,4 | 271±61,4 | 352±48,6 | 351±61,0 |
| cLDL (mg/dl) | 223±47,3 | 177±45,7 | 235±38,9 | 239±48,6 |
| cHDL (mg/dl) | 51,7±8,92 | 49,0±8,34 | 52,8±11,3 | 53,6±9,57 |
| Triglicéridos (mg/dl) | 143 [113-201] | 99,5 [82,9-163] | 178 [141-247] | 150 [116-217] |
| Apolipoproteína B (mg/dl) | 135±29,1 | 101±51,2 | 120±22,1 | 139±30,00 |
| Lipoproteína(a) (mg/dl) | 41,1 [8,48-75,4] | 72,3 [42,4-80,7] | 25,5 [15,2-34,6] | 24,4 [9,53-60,3] |
| Glucosa (mg/dl) | 89,1±11,4 | 94,6±27,3 | 95,1±12,3 | 90,5±12,4 |
| HbA1c(%) | 5,30 [5,20-5,30] | 5,30 [5,30-5,30] | 5,30 [5,30-5,30] | 5,40 [5,35-5,50] |
| Insulina (UI/ml) | 5,50 [3,58-7,60] | 4,10 [3,60-7,10] | 5,30 [4,10-7,13] | 5,70 [3,95-9,20] |
| Tratamiento con estatinas | ||||
| Ninguno | 119 (93,7) | 10 (47,6) | 0 (0) | 84 (74,3) |
| Intensidad baja | 0 | 0 | 6 (15,4) | 5 (5,95) |
| Intensidad moderada | 7 (5,52) | 10 (47,6) | 31 (79,5) | 22 (19,5) |
| Intensidad alta | 1 (0,78) | 1 (4,76) | 2 (5,13) | 2 (1,77) |
| Mutación causal en genes candidatos | ||||
| Ninguna | 114 (89,8) | 20 (95,2) | 36 (92,3) | 103 (91,2) |
| Portadores de mutación en LDLR | 8 (6,30) | 0 | 2 (5,55) | 9 (7,96) |
| Portadores de mutación en PCSK9 | 3 (2,36) | 1 (4,76) | 0 | 0 |
| Portadores de mutación en STAP1 | 2 (1,57) | 0 | 1 (2,56) | 1 (0,88) |
cHDL: colesterol unido a lipoproteínas de alta densidad; cLDL: colesterol unido a lipoproteínas de baja densidad; EC: enfermedad coronaria; HbA1c: glucohemoglobina; IMC: índice de masa corporal.
Las concentraciones de colesterol total, cLDL, cHDL y triglicéridos se han ajustado según el tratamiento con estatinas23. Algunos de los participantes pueden estar incluidos en más de un grupo.
Los valores expresan n (%), media ± desviación estándar o mediana [intervalo intercuartílico].
En el análisis de secuenciación se identificaron 5.280 variantes en genes candidatos en los 255 participantes secuenciados (figura 1): a) 1.792 variantes en el gen LDLR, 11 de las cuales se han descrito como patógenas y ya asociadas con la HF; b) 249 variantes en el gen APOB, aunque ninguna de ellas se ha descrito como patógena ni se ha asociado anteriormente con la HF; c) 1.697 variantes en el gen PCSK9, de las cuales solo 1 se ha descrito como patógena y ya asociada con la HF; d) 72 variantes en el gen APOE, 2 de las cuales ya asociadas con la hiperlipoproteinemia tipo III; e) 869 variantes en el gen STAP1, 3 de las cuales se han descrito como posiblemente patógenas mediante el análisis bioinformático, y f) 601 variantes en el gen LDLRAP1, 2 de las cuales se han clasificado como posiblemente patógenas mediante el análisis bioinformático. Sin embargo, ninguna de estas 2 variantes posiblemente patógenas del gen LDLRAP1 estaba presente en homocigosis, condición esta que se ha descrito como necesaria para causar una hipercolesterolemia.
El porcentaje de mutaciones en genes candidatos fue similar en los diversos grupos; sin embargo, los participantes con concentraciones de cLDL altas en como mínimo 2 determinaciones fueron los que presentaron un porcentaje más alto de mutaciones causales, que superó el 8% de los casos (tabla 1). Un total de 12 pacientes eran portadores de 11 alelos patógenos distintos en heterocigosis en el gen LDLR: 1 de ellos con una variante causal ubicada en la región del promotor (c.-135C>G); 7 de ellos tenían una variante causal situada en regiones codificadoras, con lo que producían un cambio de aminoácido (c.530C>T, c.826T>G, c.862G>A, c.1247G>A, c.1529C>T, c.1775G>A y c.1816G>A); 1 de ellos con una variante causal, situada en una región intrónica, que daba lugar a un cambio de corte y empalme alternativo (c.1586+5G>A); 1 alelo tenía 2 variantes causales situadas en cis: (c.274C>G) y (c.313+1G>C) y 1 de ellos tenía una reordenación grande (c.941-?_1845+?del). Todos estos casos se han descrito como patógenos mediante el análisis bioinformático y ya se habían descrito como causa de HF34-40. Tan solo 1 paciente era portador de una variante rara en el exón 26 del gen APOB, (c.10621A>G). Sin embargo, esta variante no se ha asociado anteriormente como causa de HF, se clasificó como benigna en el análisis bioinformático y no parece que afecte a la estructura de la región de unión. Se halló que 5 pacientes eran portadores de 2 variantes raras en el gen PSCK9 (c.60_65dup GCTGCT y c.743G>A), y solo la primera se ha asociado anteriormente con la HF. De estos pacientes, 4 eran portadores de un indel en el marco de lectura c.60_65dupGCTGCT, que se ha descrito como de patogenicidad incierta o probable por Clin Var41 y Garcia et al.42. También 5 pacientes eran portadores de 3 variantes raras en el gen APOE, ninguna descrita antes como causa de HF. Una de ellas nunca se ha asociado con ninguna hiperlipemia y el análisis bioinformático la clasificó como desconocida en (c.335C>A). Las otras 2 variantes (c.460C>A, c.487C>T) se hallaron en 3 y 1 participantes respectivamente. Ambas variantes ya estaban asociadas con la hiperlipoproteinemia de tipo III43,44. De los 4 pacientes portadores de 3 variantes raras desconocidas en el gen STAP1, 2 eran portadores de 1 variante situada en la región 5’ (c.-60A>G) y los otros 2 eran portadores de 2 variantes diferentes situadas en regiones de codificación, con lo que producían una sustitución de aminoácido (c.619G>A y c.803T>C). Ninguna de estas variantes se ha asociado anteriormente con la HF. Se halló que 2 pacientes eran portadores de 2 variantes raras y desconocidas en heterocigosis en el gen LDLRAP1, y 1 de ellas producía un cambio de aminoácido (c.605C>G), mientras que la otra podía afectar al corte y empalme (c.748-7C>G). Sin embargo, las mutaciones en el gen LDLRAP1 tan solo producen hipercolesterolemia cuando se encuentran en homocigosis (tabla 2 y ).
Variantes causales en genes candidatos identificadas en algunos de los pacientes de este estudio
| Gen | SNV | Nucleótido | Cambio de aminoácido | Portadores, n | Análisis bioinformático | Var. Clin. | Frecuencia ExAc32 | Frecuencia 1000 G31 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT26 | POLYPHEN-227 | Mutation Taster28 | PredictSNP29 | ||||||||
| LDLR | rs879254375 | c.-135C>G | n. p. | 1 participante | n. p. | n. p. | n. p. | n. p. | Patógena | - | - |
| LDLR | rs774467219rs112029328 | c.[274C>G;313+1G>C] | p.[Gln92Glu;NA] | 1 participante | Tolerada (0,15) | Posiblemente dañina (0,736) | Nociva(0,510) | Neutra(0,252) | Patógena | 0,0009715 | 0,001 |
| LDLR | rs121908026 | c.530C>T | p.(Ser177Leu) | 1 participante | Nociva(0,01) | Probablemente dañina (0,999) | Nociva(0,896) | Nociva (0,000090) | Patógena | 8,958e-06 | - |
| LDLR | rs879254692 | c.826T>G | p.(Cys276Gly) | 1 participante | Nociva(0) | Probablemente dañina(0,969) | Nociva(0,856) | Nociva (0,000005) | Patógena | - | - |
| LDLR | rs368657165 | c.862G>A | p.(Glu288Lys) | 1 participante | Nociva(0,05) | Probablemente dañina (0,918) | Nociva (0,714) | Nociva (0,000018) | Patógena | 0 | - |
| LDLR | - | c.941-?_1845+?del | Deleción de exón 7 a exón 12 | 1 participante | Patógena | ||||||
| LDLR | rs773658037 | c.1247G>A | p.(Arg416Gln) | 1 participante | Nociva(0,01) | Probablemente dañina (0,957) | Nociva(0,806) | Nociva (0,000045) | Patógena | 1,793e-05 | 0 |
| LDLR | rs755154048 | c.1529C>T | p.(Thr510Met) | 1 participante | Nociva (0,02) | Posiblemente dañina (0,791) | Nociva(0,679) | Nociva (0,000013) | Patógena/Probablemente patógena | 8,952e-06 | - |
| LDLR | rs781362878 | c.1586+5G>A | n. p. | 1 participante | n. p. | n. p. | n. p. | n. p. | Incierta/Patógena | 1,796e-05 | |
| LDLR | rs137929307 | c.1775G>A | p.(Gly592Glu) | 2 participantes | Nociva(0,01) | Probablemente dañina(0,925) | Nociva(0,779) | Nociva (0,000015) | Patógena | 8,951e-05 | - |
| LDLR | rs72658865 | c.1816G>A | p.(Ala606Thr) | 1 participante | Nociva(0,02) | Posiblemente dañina(0,5) | Nociva (0,550) | Nociva (0,000034) | Probablemente benigna/Incierta | 8,952e-06 | - |
| PCSK9 | rs371488778 | c.60_65dupGCTGCT | p.(Leu22_Leu23dup) | 4 participantes | n. p. | n. p. | n. p. | Nociva (0,000002) | Incierta/ Patógena | 0,002144 | - |
n. p.: no procede.
En 24 (9,41%) participantes con criterios de sospecha de HF a causa de un cLDL > percentil 95, cuando se analizó el colesterol transportado en la Lp(a), los valores de cLDL dejaron de ser > percentil 95. Ninguno de estos participantes con hiperLp(a) era portador de una mutación patógena en genes candidatos para la HF.
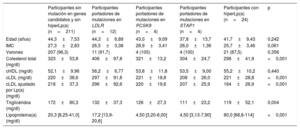
Características según el análisis genéticoEn la tabla 3 se muestran las características clínicas y el perfil lipídico según la clasificación genética, con la inclusión de los participantes con hiperLp(a). Los participantes portadores de mutaciones en el gen LDLR fueron los que mostraron las concentraciones más altas de colesterol total y cLDL. Además, los portadores de mutaciones en el gen PCSK9 presentaban unas concentraciones de colesterol total y cLDL significativamente superiores a las de los portadores de mutaciones del gen STAP1 y los clasificados como casos de hiperLp(a) (p < 0,001 y p < 0,001 respectivamente). Por otra parte, los participantes portadores de mutaciones en el gen STAP1 fueron los que mostraron las concentraciones más bajas de colesterol total y cLDL, inferiores a las de los participantes sin mutaciones en los genes candidatos.
Características clínicas y perfil lipídico según la causa principal de la hipercolesterolemia
| Participantes sin mutación en genes candidatos y sin hiperLp(a) (n=211) | Participantes portadores de mutaciones en LDLR (n=12) | Participantes portadores de mutaciones en PCSK9 (n=4) | Participantes portadores de mutaciones en STAP1 (n=4) | Participantes con hiperLp(a) (n=24) | p | |
|---|---|---|---|---|---|---|
| Edad (años) | 44,3±7,53 | 44,3±6,89 | 43,0±9,09 | 37,8±13,7 | 41,7±9,43 | 0,242 |
| IMC | 27,3±2,83 | 26,5±3,38 | 28,9±3,41 | 26,0±1,36 | 25,7±3,46 | 0,061 |
| Varones | 207 (96,3) | 11 (91,7) | 4 (100) | 4 (100) | 21 (87,5) | 0,356 |
| Colesterol total (mg/dl) | 323±53,8 | 406±97,8 | 321±13,2 | 304±24,7 | 298±41,9 | <0,001 |
| cHDL (mg/dl) | 52,1±9,96 | 56,2±6,77 | 53,8±11,8 | 53,5±9,00 | 55,2±10,2 | 0,440 |
| cLDL (mg/dl) | 220±38,6 | 297±91,8 | 221±18,8 | 208±26,0 | 221±28,8 | <0,001 |
| cLDL ajustado por Lp(a) (mg/dl) | 218±37,3 | 296±92,6 | 220±19,6 | 207±25,9 | 164±26,9 | <0,001 |
| Triglicéridos (mg/dl) | 172±80,3 | 132±37,3 | 126±27,3 | 111±23,2 | 119±52,1 | 0,004 |
| Lipoproteína(a) (mg/dl) | 20,3 [8,25-41,0] | 17,2 [13,9- 20,6] | 4,50 [3,20-6,00] | 4,50 [3,13-7,90] | 80,0 [68,8-114] | <0,001 |
cHDL: colesterol unido a lipoproteínas de alta densidad; cLDL: colesterol unido lipoproteínas de baja densidad; IMC: índice de masa corporal; Lp(a): lipoproteína(a).
El valor de p se calculó mediante la prueba ANOVA o la prueba de Kruskal-Wallis y la de la χ2, según procediera. Los valores expresan n (%), media ± desviación estándar o mediana [intervalo intercuartílico].
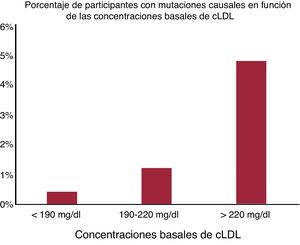
Las mutaciones en el gen LDLR implicaban al 75% de algunas mutaciones en genes candidatos, en comparación con el 25% de ellas en los participantes que eran portadores de alguna mutación en el gen PSCK9. En la figura 2 se muestra el porcentaje de mutaciones según los grupos definidos por las concentraciones de cLDL basales. Cuanto mayor es el porcentaje, mayor es el aumento del cLDL: el 0,40% de los participantes con valores basales de cLDL < 190mg/dl, el 1,20% con cLDL basal de 190-220mg/dl y un 4,80% con cLDL basal > 220 mg/dl presentaban una mutación en genes candidatos. Teniendo en cuenta a los pacientes con una mutación en genes candidatos para la HF más los pacientes con hiperLp(a), casi un 16% del grupo con criterios de sospecha tenía una enfermedad monogénica.

Se utilizó una regresión logística binaria para estudiar la asociación de los datos clínicos con 2 variables de respuesta: mutación en cualquier gen y mutación en el gen LDLR. El análisis mostró que las concentraciones basales de cLDL, el cLDL ajustado por la Lp(a) y el colesterol total tenían una asociación significativa con la presencia (sí/no) de cualquier mutación en los genes candidatos (odds ratio [OR] = 1,023; p <0,001; OR = 1,024; p < 0,001; OR = 1,017; p < 0,001) (tabla 4A-C). No obstante, ningún criterio mostró una asociación significativa con la presencia de cualquier mutación en los genes candidatos. La presencia de una mutación en el gen LDLR (sí/no) mostró una asociación significativa con las cifras basales de colesterol total y cLDL (OR = 1,009; p =0,010; OR = 1,016; p = 0,0488) y con los criterios de sospecha de HF: concentraciones de cLDL sin tratamiento superiores al percentil 95 en 1 determinación junto con una concentración de cLDL > 130mg/dl en tratamiento con estatinas (p = 0,011) (tabla 4D).
Regresión logística binaria con la presencia de mutación en genes candidatos como variable dependiente
| A | OR | IC95% | p | R2 Nagelkerke |
|---|---|---|---|---|
| Edad basal | 0,962 | 0,879-1,065 | 0,243 | 0,192 |
| IMC basal | 1,018 | 0,838-1,234 | 0,852 | |
| cLDL basal | 1,023 | 1,012-1,036 | <0,001 |
| B | OR | IC95% | p | R2 Nagelkerke |
|---|---|---|---|---|
| Edad basal | 0,958 | 0,873-1,061 | 0,379 | 0,213 |
| IMC basal | 1,026 | 0,839-1,25 | 0,797 | |
| LDL ajustadas por Lp(a) | 1,024 | 1,013-1,037 | <0,001 |
| C | OR | IC95% | p | R2 Nagelkerke |
|---|---|---|---|---|
| Edad basal | 0,961 | 0,880-1,059 | 0,389 | 0,157 |
| IMC basal | 0,995 | 0,821-1,204 | 0,959 | |
| Colesterol total basal | 1,017 | 1,008-1,025 | <0,001 |
| D | OR | IC95% | p | R2 Nagelkerke |
|---|---|---|---|---|
| Criterio 1a | 2,332 | 0,822-7,591 | 0,127 | 0,099 |
| Criterio 2b | 0,735 | 0,039-4,153 | 0,775 | |
| Criterio 3c | 0,324 | 0,026-7,607 | 0,389 | |
| Criterio 4 | 4,01 | 1,163-18,40 | 0,011 |
cLDL: colesterol unido a lipoproteínas de baja densidad; IC95%: intervalo de confianza del 95%; IMC: índice de masa corporal; LDL: lipoproteínas de baja densidad; Lp(a): lipoproteína(a); OR: odds ratio.
Participantes no tratados con concentraciones de cLDL > percentil 95 de la población española en al menos 2 determinaciones.
Participantes en tratamiento con estatinas los últimos 6 años y cLDL > 130mg/dl en al menos 2 determinaciones.
Participantes no tratados con concentraciones de cLDL > percentil 95 de la población española en 1 determinación y también cLDL > 130mg/dl en tratamiento con estatinas. A-C: regresión logística binaria utilizando la presencia de cualquier mutación (sí/no) en cualquier gen candidato. D: regresión logística binaria utilizando la presencia de cualquier mutación (sí/no) en el gen LDLR.
En la tabla 5 se muestra el número de participantes con una mutación en función de los criterios que cumplían, los valores predictivos positivo y negativo y la sensibilidad y la especificidad respecto a la muestra seleccionada por la presencia de al menos 1 criterio. El primer criterio fue el que mostró el valor predictivo positivo más alto (10,2%) para detectar mutaciones funcionales en los genes candidatos (genes LDLR,PCSK9 y STAP1), en especial para detectar mutaciones en los genes LDLR y PCSK9 (el 6,3 y el 2,4%). No obstante, el cuarto criterio fue el que mostró un valor predictivo positivo más alto para detectar mutaciones en el gen LDLR (8,0%), y mostró en esta muestra seleccionada una especificidad superior a la del primer criterio (0,510 y 0,572 respectivamente).
Número de participantes portadores de una mutación, según los criterios que cumplan
| Participantes que cumplen criterios | VPP | VPN* | Sensibilidad* | Especificidad* | ||||
|---|---|---|---|---|---|---|---|---|
| Criterio 1 | Sí (n=127) | No (n=128) | ||||||
| Mutación | Sí | No | Sí | No | ||||
| Participantes con mutación | 13 | 114 | 7 | 121 | 0,102 | 0,945 | 0,650 | 0,514 |
| Participantes con una mutación en LDLR | 8 | 119 | 4 | 124 | 0,063 | 0,969 | 0,667 | 0,510 |
| Participantes con una mutación en PCSK9 | 3 | 124 | 1 | 127 | 0,024 | 0,992 | 0,750 | 0,506 |
| Participantes con una mutación en STAP1 | 2 | 125 | 2 | 126 | 0,016 | 0,984 | 0,500 | 0,498 |
| Criterio 2 | Sí (n=21) | No (n=234) | VPP | VPN* | Sensibilidad* | Especificidad* | ||
|---|---|---|---|---|---|---|---|---|
| Mutación | Sí | No | Sí | No | ||||
| Participantes con mutación | 1 | 20 | 19 | 215 | 0,048 | 0,919 | 0,050 | 0,915 |
| Participantes con una mutación en LDLR | 0 | 21 | 12 | 222 | 0,000 | 0,949 | 0,000 | 0,914 |
| Participantes con una mutación en PCSK9 | 1 | 20 | 3 | 231 | 0,048 | 0,987 | 0,750 | 0,920 |
| Participantes con una mutación en STAP1 | 0 | 21 | 4 | 230 | 0,000 | 0,983 | 0,000 | 0,916 |
| Criterio 3 | Sí (n=39) | No (n=216) | VPP | VPN* | Sensibilidad* | Especificidad* | ||
|---|---|---|---|---|---|---|---|---|
| Mutación | Sí | No | Sí | No | ||||
| Participantes con mutación | 3 | 36 | 17 | 199 | 0,077 | 0,921 | 0,150 | 0,847 |
| Participantes con una mutación en LDLR | 2 | 37 | 10 | 206 | 0,051 | 0,954 | 0,166 | 0,848 |
| Participantes con una mutación en PCSK9 | 0 | 39 | 4 | 212 | 0,000 | 0,981 | 0,000 | 0,845 |
| Participantes con una mutación en STAP1 | 1 | 38 | 3 | 213 | 0,026 | 0,986 | 0,250 | 0,849 |
| Criterio 4 | Sí (n=113) | No (n=142) | VPP | VPN* | Sensibilidad* | Especificidad* | ||
|---|---|---|---|---|---|---|---|---|
| Mutación | Sí | No | Sí | No | ||||
| Participantes con mutación | 10 | 103 | 10 | 132 | 0,088 | 0,930 | 0,500 | 0,562 |
| Participantes con una mutación en LDLR | 9 | 104 | 3 | 139 | 0,080 | 0,979 | 0,750 | 0,572 |
| Participantes con una mutación en PCSK9 | 0 | 113 | 4 | 138 | 0,000 | 0,972 | 0,000 | 0,550 |
| Participantes con una mutación en STAP1 | 1 | 112 | 3 | 139 | 0,009 | 0,979 | 0,250 | 0,553 |
VPN: valor predictivo negativo; VPP: valor predictivo positivo.
Este estudio analiza la frecuencia de mutaciones patógenas en genes candidatos para la HF monogénica en una población con sospecha clínica de HF. De nuestros resultados se puede extraer 3 conclusiones principales. En primer lugar, la concentración de cLDL alta es el principal factor asociado con un diagnóstico genético positivo; segundo, el cLDL alto por sí solo no es lo suficientemente específico para utilizarlo en la identificación de la HF, que requiere un análisis genético, y tercero, debe incluirse la concentración de Lp(a) en el algoritmo diagnóstico de la HF. Nuestro estudio analiza a más de 4.500 individuos de la cohorte del AWHS, que corresponde a una población sana de mediana edad, y muestra que aproximadamente un 5% (255 participantes) cumplían los criterios de sospecha diagnóstica de HF, pero solo 16 (0,4%) tenían una mutación patógena en los genes LDLR o PCSK9.
La identificación de la HF es una cuestión importante, ya que los portadores de una mutación de HF presentan un aumento sustancial del riesgo de EC20, de modo que el estudio genético permite la identificación de los sujetos con hipercolesterolemia en mayor riesgo5. El análisis de los genes candidatos junto con la cuantificación de la Lp(a) permite la identificación de casi un 16% de las causas de estos grupos de hipercolesterolemia grave. Nuestros datos coinciden con los estudios publicados anteriormente sobre la prevalencia de la hiperLp(a) como causa de hipercolesterolemia primaria18,45 y refuerzan la idea de que la evaluación genética de un paciente con sospecha de HF debe incluir los genes candidatos y la cuantificación de la concentración de Lp(a)9.
El concepto de HF está evolucionando y, posiblemente, con los criterios clínicos actuales, la enfermedad sea un síndrome de hipercolesterolemia grave genética, que a veces es monogénico y otras, poligénico o de base compleja46. Este artículo hace referencia a la HF heterocigota, que es la forma más frecuente de la HF con diagnóstico clínico definitivo y la que muestra una asociación más estrecha con la ECV y, por consiguiente, requiere que se establezca más pronto un diagnóstico de presunción.
Tradicionalmente, se ha estimado que la prevalencia de la HF es de 1:5002. Sin embargo, estudios posteriores han puesto de manifiesto que la HF clínica probablemente sea más frecuente de lo que se había pensado, con una prevalencia de 1:217 en el estudio Copenhagen General Population3. En nuestro estudio, se observó que 255 participantes, de un total de 4.514 individuos estudiados, cumplían los criterios de sospecha de HF, lo cual indica que 1:18 cumplían los criterios de sospecha de HF. De estos 255 participantes, 20 tenían una variante rara en los genes LDLR,PCSK9 o STAP1, lo cual apuntaría a una prevalencia de HF de 1:226. De un total de 20 participantes con variantes raras en genes candidatos, 12 tenían una mutación en el gen LDLR, 4 la tenían en el gen PCSK9 y 4 tenían una variante en el gen STAP1.
Según lo indicado por estudios recientes16,17,47, el papel del gen STAP1 no se ha asociado claramente con el fenotipo de HF y, por este motivo, nosotros recalculamos la prevalencia de la HF considerando solo a los portadores de mutaciones en los genes LDLR y PCSK9. Con este criterio, la prevalencia disminuiría de 1:226 a 1:282. En el presente estudio, hemos puesto de manifiesto que los portadores de LDLR FH presentan el fenotipo de HF extrema, tal como se había descrito anteriormente48,49. Por consiguiente, si tenemos en cuenta tan solo a los portadores del gen LDLR, la prevalencia de la HF se reduce de 1:226 a 1:376.
En el análisis de secuenciación de los genes LDLR, APOB, PCSK9, APOE, STAP1 y LDLRAP1, se identificaron 5.280 variantes, pero solo 16 de ellas fueron posiblemente patógenas o patógenas según el análisis in silico:a) 11 variantes patógenas en el gen LDLR que ya estaban asociadas con la HF34-40; b) 1 variante patógena en el gen PCSK9 (c.60_65dupGCTGCT), que tiene una frecuencia < 0,5% de la población general y se ha clasificado como patógena mediante el análisis bioinformático; sin embargo, serán necesarios nuevos estudios para investigar la funcionalidad de esta variante; c) 1 variante rara en el gen APOB (c.10621A>G); esta variante que produce el cambio de aminoácido p.(Ile3515Val) en la proteína madura está situado en el dominio beta 2 de la apolipoproteína B, y las herramientas de predicción computacionales y los análisis de conservación indican que esta variante no tendrá repercusión en la función de la proteína; además, las herramientas de ayuste computacionales indican que esta variante no tendría repercusiones en el ayuste del ARN50; d) 3 variantes raras en el gen STAP1, 1 de ellas situada en el 5’UTR (c.-60A>G) y 2 en regiones de codificación (c.619G>A y c.803T>C) que producen cada una un cambio de aminoácido, es decir, p.(Ile268Thr) y p.(Asp207Asn) respectivamente; todas estas variantes se han clasificado como patógenas mediante el análisis bioinformático, pero de ninguna de ellas se ha establecido antes una asociación con la HF, y serán necesarios más estudios sobre el papel del gen STAP1 en el fenotipo de la HF, ya que en estudios previos se ha descrito a participantes que son portadores de mutaciones del gen STAP1 y presentan unas concentraciones normales de colesterol total y cLDL y una asociación incompleta con la HF16,17,51; e) 2 variantes raras en el gen LDLRAP1, 1 de ellas situada en la región intrónica (c.748-7C>G), que podría producir un ayuste alternativo, y otra situada en una región de codificación (c.605C>G), que conllevaría el cambio de aminoácido p.(Ser202Cys); esta variante se ha clasificado como patógena mediante el análisis bioinformático, pero el participante era portador de esta variante en heterocigosis, y ello no explicaría el fenotipo de HF, y f) 3 variantes missense raras en el gen APOE, 2 de ellas (c.460C>A y c.487C>T) descritas como patógenas por Clin Var52 y asociadas a la hiperlipoproteinemia de tipo III43,44, pero no a la HF.
Nuestros resultados muestran una asociación significativa de las concentraciones de cLDL con la presencia de una mutación causal en genes candidatos. Aunque en nuestra muestra, procedente de una población laboral, la EC fue escasa y mostró el más bajo valor predictivo positivo de las mutaciones, los participantes con cifras de cLDL sin tratamiento > 220mg/dl y aquellos con cLDL > 130mg/dl a pesar del tratamiento con estatinas mostraron una asociación significativa con la presencia de una mutación patógena. En ellos, la probabilidad de hallar una mutación causal en un gen candidato fue 4 veces superior a la de los pacientes que cumplían los demás criterios de sospecha de HF. En el futuro, la disminución progresiva de los costes del análisis del ADN probablemente facilite la aplicación de programas universales de detección sistemática en la población. Sin embargo, dados sus costes actuales, se hace necesario seleccionar a los candidatos a las pruebas genéticas. Nuestros resultados indican que no solo las altas concentraciones de cLDL sin tratamiento son un buen marcador de la HF en la población general, sino que la combinación de estos valores con concentraciones altas de cLDL a pesar del tratamiento con estatinas podría ser un mejor predictor de la HF. Además, los participantes con cifras de cLDL altas sin tratamiento y cifras altas pese al tratamiento con estatinas fueron los que mostraron mayores porcentajes de mutaciones en LDLR (7,96%), y estos participantes mostraron también un fenotipo de HF más extremo.
El porcentaje de mutaciones en genes candidatos muestra grandes diferencias, con valores que van del 55,6% cuando se dispone de información de antecedentes de un patrón monogénico familiar de hipercolesterolemia8 a menos del 2% si se usa exclusivamente la concentración de LDL > 190mg/dl20. Nuestro estudio es plenamente coherente con el concepto de que una concentración de cLDL alta por sí sola, sin información de antecedentes familiares de hipercolesterolemia grave, permite la identificación de un pequeño porcentaje de pacientes con HF.
LimitacionesNuestro estudio tiene ciertas limitaciones. En primer lugar, el AWHS es una cohorte con grandes prevalencias de varones, obesidad, hipertensión e hipercolesterolemia, y ello puede hacer que no sea representativa de la población general. Sin embargo, dado que se trata de una población relativamente joven, no habrá un sesgo en la prevalencia de mutaciones de HF. En segundo lugar, se presentan algunas mutaciones, como la c.743G>A en PCSK9 y la c.10621A>G en APOB, que tienen una significación incierta. No obstante, ambas se han descrito como neutras o benignas mediante el análisis bioinformático y la base de datos ClinVar, por lo que no hay datos suficientes para clasificarlas como variantes patógenas. Los análisis de secuenciación del gen APOB incluyeron solo los exones 26 y 29, mientras que la secuenciación del gen APOB completo podría mostrar otras variantes como causa del fenotipo de HF. Sin embargo, la hipercolesterolemia debida a defectos del gen APOB requeriría que la proteína tuviera una unión defectuosa con el receptor de LDL, ya que las mutaciones que causan solo una menor expresión o un plegado inadecuado de la ApoB causarían hipocolesterolemia. Así pues, es de prever que las variantes causantes de HF estén situadas en la región de codificación del dominio de unión de la ApoB (en los exones 26 y 29). De hecho, la mayoría de las mutaciones del gen APOB descritas como causa de HF se han identificado en el exón 2649. Por último, las mutaciones posiblemente patógenas de PCSK9 (c.60_65dupGCTGCT) y STAP1 (c.-60A>G, c.619G>A y c.803T>C) requerirán nuevos estudios para disponer de una perspectiva más amplia respecto a su papel en el fenotipo de HF.
CONCLUSIONESEl presente estudio analiza el valor predictivo positivo de los criterios diagnósticos clínicos para la HF. En nuestro estudio se incluyó a un total de 4.514 individuos de la cohorte del AWHS, formada por una población de mediana edad sana. Cumplían los criterios diagnósticos para la sospecha clínica de HF 255 individuos, y 16 de ellos (6,27%) presentaban mutaciones en los genes LDLR y PCSK9, lo cual corresponde a una prevalencia de HF de 1:282 en esta población. Además, a 24 participantes (9,41%) se les diagnosticó una hiperLp(a), lo cual permitió identificar casi un 16% de la etiología de estos grupos de hipercolesterolemia grave. Las concentraciones de cLDL sin tratamiento y los valores de cLDL altos a pesar del tratamiento con estatinas mostraron una asociación significativa con la presencia de mutaciones en los genes candidatos. Estos resultados indican que la combinación de un cLDL alto sin tratamiento y valores de cLDL altos a pesar del tratamiento con estatinas podría ser un primer paso, aunque no el único, para el cribado poblacional de la HF.
FINANCIACIÓNEste trabajo contó con el apoyo de subvenciones del Gobierno de Aragón, B14-7R, España y del Ministerio de Economía y Competitividad de España PI15/01983, PI18/01777 y CIBERCV. Estos proyectos son cofinanciados por el Instituto de Salud Carlos III y el Fondo Europeo de Desarrollo Regional (FEDER) de la Unión Europea «Una manera de hacer Europa». CIBERCV es un proyecto del Instituto de Salud Carlos III.
CONFLICTO DE INTERESESLos autores declaran no tener conflictos de intereses.
- –
La HF es un trastorno genético caracterizado por altas concentraciones plasmáticas de colesterol total y cLDL y alto riesgo de ECV.
- –
Se ha demostrado que los criterios clínicos muestran una gran asociación con el diagnóstico genético.
- –
Sin embargo, un 20-40% de los pacientes con el fenotipo de HF no presentan una mutación en los genes candidatos.
- –
La European Atherosclerosis Society ha recomendado emplear unos nuevos criterios diagnósticos para la sospecha de HF, que no han sido validados aún.
- –
La prevalencia de la HF en España es de 1:282.
- –
Tan solo un 6,27% de los participantes con sospecha de HF presentaban mutaciones funcionales en los genes LDLR y PCSK9.
- –
A un total del 9,41% de los participantes con sospecha de HF se les diagnosticó una hiperlipoproteinemia(a).
- –
La combinación de una concentración de cLDL alta sin tratamiento y valores de cLDL altos a pesar del tratamiento con estatinas es el mejor predictor de un resultado positivo para una mutación de HF.