La miocardiopatía arritmogénica del ventrículo derecho (MCAVD) es una cardiopatía hereditaria definida por la sustitución progresiva de miocardio ventricular derecho por tejido fibroadiposo. Es causa frecuente de la muerte súbita de jóvenes atletas. El objetivo del presente estudio es conocer la incidencia de variantes desmosómicas patogénicas o probablemente patogénicas en pacientes con MCAVD definitiva de alto riesgo.
MétodosEl estudio de cohortes retrospectivo observacional incluyó a 36 pacientes diagnosticados de MCAVD definitiva de alto riesgo en nuestro hospital entre enero de 1998 y enero de 2015. El análisis genético se realizó con next-generation sequencing.
ResultadosLa mayoría eran varones (28 pacientes, 78%) con una media de edad al diagnóstico de 45 ± 18 años. Se detectó al menos 1 variante desmosómica patogénica o probablemente patogénica en 26 de los 35 casos índice (74%): 5 nonsense, 14 frameshift, 1 splice y 6 missense. En 15 pacientes (71%) se encontraron mutaciones nuevas. La presencia o la ausencia de mutaciones desmosómicas o la naturaleza de estas no se asociaron con características electrocardiográficas, clínicas, arrítmicas, anatómicas o pronósticas específicas.
ConclusionesLa incidencia de variantes desmosómicas patogénicas o probablemente patogénicas en MCAVD definitiva de alto riesgo fue muy alta, con mayoría de mutaciones que causan truncamiento. La presencia de mutaciones desmosómicas no se asoció con el pronóstico.
Palabras clave
La miocardiopatía arritmogénica del ventrículo derecho (MCAVD) es una cardiopatía definida por la sustitución progresiva de miocardio ventricular derecho por tejido fibroadiposo, que puede dar lugar a arritmias, muerte súbita e insuficiencia cardiaca. En la progresión de la enfermedad, suele afectarse también el ventrículo izquierdo, lo que confiere peor pronóstico. Se hereda habitualmente de modo autosómico dominante por mutaciones en genes que codifican proteínas desmosómicas. Actualmente, en torno al 40-60% de los casos clínicamente diagnosticados muestran al menos 1 variación genética que causa la enfermedad1–8.
Aún no se dispone de ensayos aleatorizados prospectivos sobre el uso del desfibrilador automático implantable (DAI) en pacientes con MCAVD, y los predictores independientes de eventos arrítmicos provienen de estudios retrospectivos9–11. Se ha desarrollado un documento de consenso para el tratamiento de esta enfermedad, que divide a los pacientes en 3 categorías de riesgo: alta, intermedia y baja12. La categoría de alto riesgo incluye a los pacientes que han sufrido una parada cardiaca debida a taquicardia ventricular sostenida o fibrilación ventricular y pacientes con disfunción grave del ventrículo derecho (área fraccional ≤ 17% o fracción de eyección ≤ 35%) o disfunción grave del ventrículo izquierdo (fracción de eyección del ventrículo izquierdo ≤ 35%), incluso en ausencia de arritmias ventriculares potencialmente mortales. Se recomienda el implante de DAI para los pacientes MCAVD de alto riesgo (clase I)12.
El objetivo del presente estudio es analizar la incidencia de variantes desmosómicas patológicas o probablemente patológicas en 36 pacientes con MCAVD definitiva de alto riesgo.
MÉTODOSDiseño del estudio y poblaciónEstudio observacional de cohortes retrospectivo en el que se incluyó a los 36 pacientes con MCAVD definitiva de alto riesgo portadores de DAI con seguimiento en nuestro hospital entre enero de 2000 y enero de 2016.
El diagnóstico de MCAVD definitivo se estableció según los criterios de la Task Force modificados en 20104, si se cumplían al menos 2 criterios mayores o 1 criterio mayor y 2 menores o 4 criterios menores de categorías diagnósticas diferentes.
El criterio de alto riesgo se estableció según el documento de consenso de las sociedades internacionales de cardiología12, es decir, pacientes en prevención secundaria por taquiarritmias ventriculares o parada cardiaca previa o en prevención primaria por disfunción sistólica ventricular grave derecha y/o izquierda.
Se recogieron las características demográficas, clínicas, anatómicas, electrocardiográficas y arrítmicas, así como el número y el tipo de criterios diagnósticos que se cumplían en el momento del diagnóstico. Las características demográficas se registraron en el diagnóstico de la enfermedad. Las anatómicas, mediante ecocardiografía-Doppler o por resonancia magnética cardiaca.
El primer evento arrítmico mayor se definió como la primera taquicardia ventricular sostenida, muerte súbita recuperada o terapia apropiada en un paciente que no había sufrido antes evento alguno (aquellos con DAI en prevención primaria).
El estudio cumple la normativa ética de la Declaración de Helsinki de 1975.
GenéticaEl análisis genético se llevó a cabo en los casos índice mediante técnicas de secuenciación masiva next-generation sequencing entre diciembre de 2013 y junio de 2016 e incluyó todos los genes relacionados con la enfermedad y también con otras miocardiopatías. Los genes estudiados fueron los siguientes: DSC2, DSG2, DSP, FLNC, JUP, PKP2, PLN, TMEM43, CTNNA3, DES, LMNA, RYR2, TGFB3, TTN, CASQ2, CTNNB1, LDB3, PERP, PKP4, PPP1R13L y SCN5A. Al único familiar del estudio se le realizó secuenciación en ambas direcciones del fragmento del gen (PKP2 en este caso) de la variante genética previamente identificada.
Los exones y límites intrónicos se capturaron utilizando una biblioteca de sondas (SureSelect Target Enrichment Kit for Illumina, Agilent Technologies). La secuenciación se realizó utilizando la plataforma Illumina HiSeq 1500 (Illumina; San Diego, California, Estados Unidos) con una longitud de lectura de 2 × 100 bases siguiendo los protocolos de Illumina. El análisis bioinformático se llevó a cabo a través de diferentes programas para la genotipificación. Se consideró la información relativa a la frecuencia en diferentes poblaciones (1000 Genomes Project, Exome Variant Server, Exome Aggregation Consortium). Para el filtrado de variantes, se utilizó una estrategia interna de filtrado y priorización de variantes diseñada por Health in Code, y el análisis se concentró en los principales genes incluidos en el panel de cardiomiopatías arritmogénicas. Un equipo de cardiólogos y genetistas evaluó y clasificó las variantes mediante el esquema de clasificación de variantes Health in Code adaptado del American College of Medical Genetics.
Las variantes consideradas clínicamente relevantes según el fenotipo del paciente se confirmaron mediante secuenciación de Sanger.
SeguimientoLos eventos apropiados se definieron como la intervención del DAI mediante terapia antitaquicárdica o por choques en respuesta a arritmia ventricular sostenida. Los eventos inapropiados se definieron como la intervención del DAI con terapia antitaquicárdica o choque por taquicardia supraventricular (fibrilación auricular, taquicardia auricular o sinusal) o por disfunción del dispositivo (sobresensado o ruido).
Durante el seguimiento se recogieron los eventos arrítmicos ventriculares apropiados (número, tiempo desde el implante, longitud de ciclo, eficacia de la terapia antitaquicárdica y choques) e inapropiados (número, tiempo desde el implante y causas), la aparición de fibrilación auricular o insuficiencia cardiaca, la necesidad de ablación de sustrato, el daño en el ventrículo izquierdo y los fallecimientos o los trasplantes ocurridos.
Análisis estadísticoPara el análisis de los datos se utilizó el programa SPSS (versión 18.0). Las variables continuas se expresan como media ± desviación típica; las variables categóricas, como valor absoluto y porcentaje. Para la comparación de variables continuas, se utilizó el test de la t de Student (en caso de distribución normal) o la de la U de Mann-Whitney (para las de distribución no normal). Las variables categóricas se compararon mediante tablas de contingencia y la prueba de la χ2 o el test exacto de Fisher. El estudio del tiempo libre de eventos arrítmicos apropiados se llevó a cabo mediante el método de Kaplan-Meier.
Se consideró significación estadística con valores de p < 0,05 en sentido bilateral.
RESULTADOSCaracterísticas basales de la muestraLa mayoría eran varones (28 pacientes; 78%), con una edad media al diagnóstico de 45 ± 18 años. Todos tenían MCAVD definitiva según los criterios de Task Force modificados en 20104, independientemente del resultado genético.
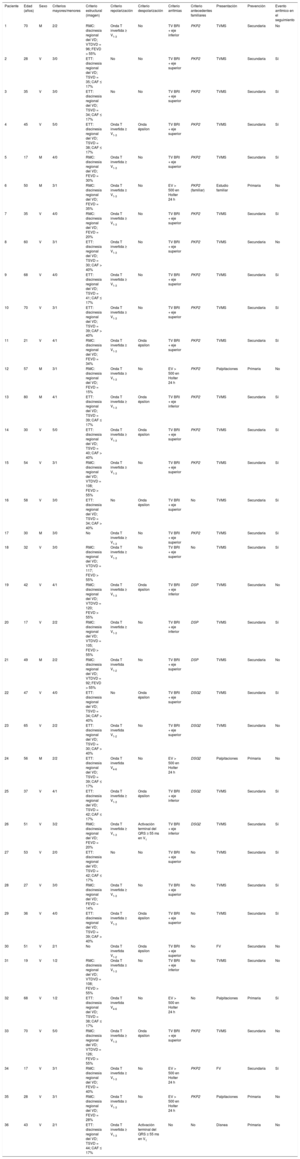
Los criterios diagnósticos y las características clínicas principales se desglosan en la tabla 1, teniendo en cuenta que a ningún paciente se le realizó biopsia endomiocárdica para establecer el diagnóstico, aunque el de un paciente se confirmó a posteriori porque se le realizó trasplante cardiaco.
Criterios de la Task Force modificados de 2010 y características principales en los 36 pacientes de la muestra
| Paciente | Edad (años) | Sexo | Criterios mayores/menores | Criterio estructural (imagen) | Criterio repolarización | Criterio despolarización | Criterio arritmias | Criterio antecedentes familiares | Presentación | Prevención | Evento arrítmico en el seguimiento |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 70 | M | 2/2 | RMC: discinesia regional del VD; VTDVD = 96; FEVD > 55% | Onda T invertida ≥ V1-3 | No | TV BRI + eje inferior | PKP2 | TVMS | Secundaria | No |
| 2 | 28 | V | 3/0 | ETT: discinesia regional del VD; TSVD = 35; CAF ≤ 17% | No | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 3 | 35 | V | 3/0 | ETT: discinesia regional del VD; TSVD = 34; CAF ≤ 17% | No | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 4 | 45 | V | 5/0 | ETT: discinesia regional del VD; TSVD = 38; CAF ≤ 17% | Onda T invertida ≥ V1-3 | Onda épsilon | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 5 | 17 | M | 4/0 | RMC: discinesia regional del VD; FEVD = 30% | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 6 | 50 | M | 3/1 | RMC: discinesia regional del VD; FEVD = 35% | Onda T invertida ≥ V1-3 | No | EV > 500 en Holter 24 h | PKP2 (familiar) | Estudio familiar | Primaria | No |
| 7 | 35 | V | 4/0 | RMC: discinesia regional del VD; FEVD = 20% | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 8 | 60 | V | 3/1 | ETT: discinesia regional del VD; TSVD = 30; CAF > 40% | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | No |
| 9 | 68 | V | 4/0 | ETT: discinesia regional del VD; TSVD = 41; CAF ≤ 17% | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 10 | 70 | V | 3/1 | ETT: discinesia regional del VD; TSVD = 39; CAF > 40% | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 11 | 21 | V | 4/1 | RMC: discinesia regional del VD; FEVD = 34% | Onda T invertida ≥ V1-3 | Onda épsilon | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 12 | 57 | M | 3/1 | RMC: discinesia regional del VD; FEVD = 15% | Onda T invertida ≥ V1-3 | No | EV > 500 en Holter 24 h | PKP2 | Palpitaciones | Primaria | No |
| 13 | 80 | M | 4/1 | ETT: discinesia regional del VD; TSVD = 39; CAF ≤ 17% | Onda T invertida ≥ V1-3 | Onda épsilon | TV BRI + eje inferior | PKP2 | TVMS | Secundaria | Sí |
| 14 | 30 | V | 5/0 | ETT: discinesia regional del VD; TSVD = 40; CAF > 40% | Onda T invertida ≥ V1-3 | Onda épsilon | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 15 | 54 | V | 3/1 | RMC: discinesia regional del VD; VTDVD = 108; FEVD > 55% | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 16 | 58 | V | 3/0 | ETT: discinesia regional del VD; TSVD = 34; CAF > 40% | No | Onda épsilon | TV BRI + eje superior | No | TVMS | Secundaria | Sí |
| 17 | 30 | M | 3/0 | No | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | PKP2 | TVMS | Secundaria | Sí |
| 18 | 32 | V | 3/0 | RMC: discinesia regional del VD; VTDVD = 117; FEVD > 55% | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | No | TVMS | Secundaria | Sí |
| 19 | 42 | V | 4/1 | RMC: discinesia regional del VD; VTDVD = 120; FEVD > 55% | Onda T invertida ≥ V1-3 | Onda épsilon | TV BRI + eje inferior | DSP | TVMS | Secundaria | No |
| 20 | 17 | V | 2/2 | RMC: discinesia regional del VD; VTDVD = 105; FEVD > 55% | Onda T invertida ≥ V1-3 | No | TV BRI + eje inferior | DSP | TVMS | Secundaria | Sí |
| 21 | 49 | M | 2/2 | RMC: discinesia regional del VD; VTDVD = 92; FEVD > 55% | Onda T invertida V1-2 | No | TV BRI + eje superior | DSP | TVMS | Secundaria | No |
| 22 | 47 | V | 4/0 | ETT: discinesia regional del VD; TSVD = 34; CAF > 40% | No | Onda épsilon | TV BRI + eje superior | DSG2 | TVMS | Secundaria | Sí |
| 23 | 65 | V | 2/2 | ETT: discinesia regional del VD; TSVD = 30; CAF > 40% | Onda T invertida V1-2 | No | TV BRI + eje superior | DSG2 | TVMS | Secundaria | No |
| 24 | 56 | M | 2/2 | ETT: discinesia regional del VD; TSVD = 39; CAF ≤ 17% | Onda T invertida V4-6 | No | EV > 500 en Holter 24 h | DSG2 | Palpitaciones | Primaria | No |
| 25 | 37 | V | 4/1 | ETT: discinesia regional del VD; TSVD = 42; CAF ≤ 17% | Onda T invertida ≥ V1-3 | Onda épsilon | TV BRI + eje inferior | DSG2 | TVMS | Secundaria | Sí |
| 26 | 51 | V | 3/2 | RMC: discinesia regional del VD; FEVD = 20% | Onda T invertida ≥ V1-3 | Activación terminal del QRS ≥ 55 ms en V1 | TV BRI + eje inferior | DSG2 | TVMS | Secundaria | Sí |
| 27 | 53 | V | 2/0 | ETT: discinesia regional del VD; TSVD = 42; CAF ≤ 17% | No | No | TV BRI + eje superior | No | TVMS | Secundaria | Sí |
| 28 | 27 | V | 3/0 | RMC: discinesia regional del VD; FEVD = 14% | Onda T invertida ≥ V1-3 | No | TV BRI + eje superior | No | TVMS | Secundaria | Sí |
| 29 | 36 | V | 4/0 | ETT: discinesia regional del VD; TSVD = 39; CAF > 40% | Onda T invertida ≥ V1-3 | Onda épsilon | TV BRI + eje superior | No | TVMS | Secundaria | Sí |
| 30 | 51 | V | 2/1 | No | Onda T invertida V1-2 | Onda épsilon | TV BRI + eje superior | No | FV | Secundaria | No |
| 31 | 19 | V | 1/2 | RMC: discinesia regional del VD; VTDVD = 108; FEVD > 55% | Onda T invertida ≥ V1-3 | No | TV BRI + eje inferior | No | TVMS | Secundaria | No |
| 32 | 68 | V | 1/2 | ETT: discinesia regional del VD; TSVD = 38; CAF ≤ 17% | Onda T invertida V4-6 | No | EV > 500 en Holter 24 h | No | Palpitaciones | Primaria | Sí |
| 33 | 70 | V | 5/0 | RMC: discinesia regional del VD; VTDVD = 126; FEVD > 55% | Onda T invertida ≥ V1-3 | Onda épsilon | TV BRI + eje superior | PKP2 | TVMS | Secundaria | No |
| 34 | 17 | V | 3/1 | RMC: discinesia regional del VD; FEVD = 40% | Onda T invertida ≥ V1-3 | No | EV > 500 en Holter 24 h | PKP2 | FV | Secundaria | Sí |
| 35 | 28 | V | 3/1 | RMC: discinesia regional del VD; FEVD = 28% | Onda T invertida ≥ V1-3 | No | EV > 500 en Holter 24 h | PKP2 | Palpitaciones | Primaria | No |
| 36 | 43 | V | 2/1 | ETT: discinesia regional del VD; TSVD = 44; CAF ≤ 17% | Onda T invertida ≥ V1-3 | Activación terminal del QRS ≥ 55 ms en V1 | No | No | Disnea | Primaria | No |
CAF: cambio de área fraccional; DSG2: desmogleína 2; DSP: desmoplaquina; ETT: ecocardiografía transtorácica; EV: extrasistolia ventricular; FEVD: fracción de eyección del ventrículo derecho; FV: fibrilación ventricular; M: mujeres; PKP2: plakofilina 2; RMC: resonancia magnética cardiaca; TSVD: tracto de salida del ventrículo derecho en proyección paraesteral de eje largo; TV BRI: taquicardia ventricular con morfología de bloqueo de rama izquierda; TVMS: taquicardia ventricular monomorfa sostenida; V: varón; VD: ventrículo derecho; VTDVD: volumen telediastólico del VD indexado por superficie corporal (ml/m2).
Respecto a la presentación de la enfermedad, en la mayoría se diagnosticó a partir de eventos arrítmicos: 2 pacientes tuvieron muerte súbita recuperada y 28, taquicardia ventricular sostenida con morfología de bloqueo de rama izquierda (20 de eje superior y 8 de eje inferior); se diagnosticó a 4 de ellos a partir de estudio por extrasistolia ventricular, a 1 durante el estudio con insuficiencia cardiaca y a otro durante el cribado familiar (único familiar del estudio, madre de caso índice mujer). A los primeros 30 pacientes, se les implantó DAI en prevención secundaria y a los 6 restantes, en prevención primaria por tener daño ventricular grave (2 biventricular y 4 ventricular derecha).
La media de fracción de eyección del ventrículo izquierdo fue del 58 ± 10%; 6 pacientes (17%) tenían daño del ventrículo izquierdo y 2 (6%) presentaron disfunción ventricular grave (fracción de eyección del ventrículo izquierdo < 35%). La media de FE del ventrículo derecho fue del 40 ± 18% medida por resonancia cardiaca, y 18 pacientes (50%) tenían disfunción grave del ventrículo derecho (fracción de eyección del ventrículo derecho < 35%) medida por ecocardiografía o resonancia.
Se halló realce tardío con gadolinio en 10 de los 18 pacientes a los que se hizo resonancia cardiaca: en 7 pacientes en ventrículo derecho; en 2, biventricular y en 1, exclusivamente en el ventrículo izquierdo.
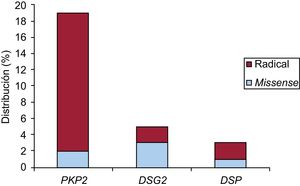
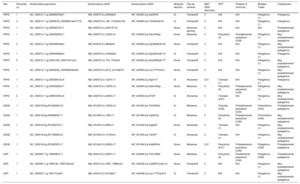
GenéticaMediante next-generation sequencing se detectó al menos 1 variante desmosómica patogénica o probablemente patogénica en 27 pacientes (75%): 26 casos índice y 1 familiar. De ellas, las de la plakofilina 2 (PKP2) fueron las más frecuentes, con 19 variantes, seguidas de las 5 en la desmogleína 2 (DSG2) y las 3 de la desmoplakina (DSP).
Respecto a la naturaleza, había 5 nonsense, 15 frameshifty, 1 splice y 6 missense. En 15 pacientes (71%) se obtuvieron 10 mutaciones nuevas, no descritas previamente. Una de ellas (NP_004563.2:p.Glu259Glyfs*77) se detectó en 5 casos índice de 5 familias diferentes en la misma provincia; en otra (NP_004563.2: p.Gly328Glufs*24) hubo cosegregación y además un familiar cumplía criterios de alto riesgo (disfunción ventricular derecha), por lo que se lo incluyó en el estudio (único familiar). Respecto a las 8 variantes restantes, se estudió a 49 familiares, y se detectó a 21 portadores y cosegregación en 5 de las variantes, pero ningún familiar cumplía los criterios de MCAVD de alto riesgo.
En la figura 1 se puede ver su distribución en función del gen afectado y su naturaleza. Sus características se desglosan en la tabla 2.

Variantes desmosómicas de los pacientes del estudio
| Gen | Pacientes (n) | Nomenclatura genómica | Nomenclatura cADN | Nomenclatura cADN | Mutación descrita | Tipo de variante | MAF 5000 Genomes | SIFT | Poliphen-2 (HumVar) | Mutation Taster | Clasificación |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PKP2 | 1 | NC_000012.11:g.32945647delT | NM_004572.3:c.2509delA | NP_004563.2:p.Ser837fs | Sí | Frameshift | 0 | N/A | N/A | Patogénica (1) | Patogénica |
| PKP2 | 3 | NC_000012.11:g.33049518_33049521delTCTG | NM_004572.3:c.148_151delACAG | NP_004563.2:p.Thr50Serfs*61 | Sí | Frameshift | 0 | N/A | N/A | Patogénica (1) | Patogénica |
| PKP2 | 1 | NC_000012.11:g.32949237A>T | NM_004572.3:c.2300-5T>A | Novel | Abnormal splicing | 0 | N/A | N/A | N/A | Probablemente patogénica | |
| PKP2 | 1 | NC_000012.11:g.33003825G>T | NM_004572.3:c.1253C>A | NP_004563.2:p.Ala418Asp | Novel | Missense | 0 | Perjudicial (0) | Probablemente perjudicial (0,99) | Patogénica (1) | Muy probablemente patogénica |
| PKP2 | 2 | NC_000012.11:g.33030835delC | NM_004572.3:c.983delG | NP_004563.2:p.Gly328Glufs*24 | Novel | Frameshift | 0 | N/A | N/A | Patogénica (1) | Muy probablemente patogénica |
| PKP2 | 1 | NC_000012.11:g.32994008delC | NM_004572.3:c.1643delG | NP_004563.2:p.Gly548Valfs*15 | Sí | Frameshift | 0 | N/A | N/A | Patogénica (1) | Patogénica |
| PKP2 | 5 | NC_000012.11:g.33031040_33031041insC | NM_004572.3:c.775_776insG | NP_004563.2:p.Glu259Glyfs*77 | Novel | Frameshift | 0 | N/A | N/A | Patogénica (1) | Muy probablemente patogénica |
| PKP2 | 1 | NC_000012.11:g.32949222_32949223delAG | NM_004572.3:c.2312_2313delTC | NP_004563.2:p.Leu771Profs*2 | Novel | Frameshift | 0 | N/A | N/A | Patogénica (1) | Muy probablemente patogénica |
| PKP2 | 1 | NC_000012.11:g.33003841G>A | NM_004572.3:c.1237C>T | NP_004563.2:p.Arg413* | Sí | Nonsense | 0,01 | Tolerada (1) | N/A | Patogénica (1) | Patogénica |
| PKP2 | 1 | NC_000012.11:g.33003825G>T | NM_004572.3:c.1253C>A | NP_004563.2:p.Ala418Asp | Novel | Missense | 0 | Perjudicial (0) | Probablemente perjudicial (0,99) | Patogénica (1) | Muy probablemente patogénica |
| PKP2 | 2 | NC_000012.11:g.32955433G>A | NM_004572.3:c.2203C>T | NP_004563.2:p.R735* | Sí | Nonsense | 0 | Tolerada (0,87) | N/A | Patogénica (1) | Muy probablemente patogénica |
| DSG2 | 1 | NC_000018.9:g.29104840A>G | NM_001943.3:c.1003A>G | NP_001934.2:p.Thr335Ala | Sí | Missense | 0 | Tolerada (0,08) | Probablemente perjudicial (0,94) | Polimorfismo (0,52) | Probablemente patogénica |
| DSG2 | 1 | NC_000018.9:g.29099820C>T | NM_001943.3:c.136C>T | NP_001934.2:p.Arg46Trp | Sí | Missense | 0 | Perjudicial (0) | Probablemente perjudicial (0,99) | Polimorfismo (0,99) | Muy probablemente patogénica |
| DSG2 | 1 | NC_000018.9:g.29102207A>T | NM_001943.3:c.685A>T | NP_001934.2:p.Arg229* | Novel | Nonsense | 0 | Tolerada (1) | N/A | Patogénica (1) | Muy probablemente patogénica |
| DSG2 | 1 | NC_000018.9:g.29115262G>A | NM_001943.3:c.1310G>A | NP_001934.2:p.Trp437* | Sí | Nonsense | 0 | Tolerada (1) | N/A | Patogénica (1) | Muy probablemente patogénica |
| DSG2 | 1 | NC_000018.9:g.29126255C>T | NM_001943.3:c.2906C>T | NP_001934.2:p.Ala969Val | Novel | Missense | 0,01 | Perjudicial (0,01) | Probablemente perjudicial (0,98) | Patogénica (0,96) | Probablemente patogénica |
| DSP | 1 | NC_000006.11:g.7584082C>T | NM_004415.2:c.6587C>T | NP_004406.2:p.Thr2196Ile | Novel | Missense | 0 | Perjudicial (0) | Posiblemente perjudicial (0,90) | Patogénica (0,96) | Probablemente patogénica |
| DSP | 1 | NC_000006.11:g.7585192_7585193insG | NM_004415.2:c.7697_7698insG | NP_004406.2:p.Val2567Cysfs*14 | Novel | Frameshift | 0 | N/A | N/A | Patogénica (1) | Muy probablemente patogénica |
| DSP | 1 | NC_000006.11:g.7581741delT | NM_004415.2:c.5318delT | NP_004406.2:p.Leu1773Tyrfs*8 | Sí | Frameshift | 0 | N/A | N/A | Patogénica (1) | Muy probablemente patogénica |
DSG2: desmogleína 2; DSP: desmoplaquina; N/A: no aplicable PKP2: plakofilina 2.
Además, se encontraron variantes desmosómicas clasificadas como probablemente no patológicas en 3 pacientes: 2 afectados de proteína plakoglobina (JUP) (NP_068831.1:p.Lys261Trpfs*39 y NP_068831.1:p.Glu146Lys) y otro en la desmocolina 2 (DSC2) (abnormal splicing intron 9).
En 4 pacientes se detectaron, además de las mutaciones desmosómicas, otras variantes en genes no desmosómicos. En concreto, 3 en el gen que codifica la titina (TTN) y 1 en el gen del receptor de la rianodina (RYR2), lo que no se asoció con ninguna característica clínica o pronóstica determinada. No se detectaron variantes aisladas en genes no desmosómicos relacionados con la enfermedad.
La mutación del gen PKP2 se asoció con un daño exclusivamente en el ventrículo derecho (el 100% de daño el ventrículo derecho frente a un 0% en el izquierdo o biventricular; p < 0,05). Respecto a los 6 pacientes con daño del ventrículo izquierdo, 2 tenían mutación de la DSP (el 67 frente al 33%; p = 0,07), 1 mutación de la DSG2 y en los últimos 3 el test genético resultó negativo.
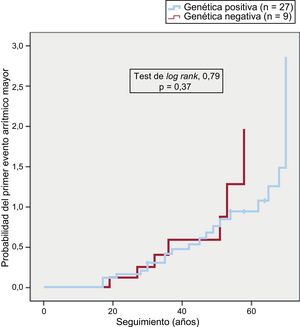
La presencia de variantes patogénicas, el tipo de gen afectado o la naturaleza de aquellas no se asociaron con un primer evento arrítmico más precoz (figura 2), una presentación clínica diferente o determinadas características electrocardiográficas y anatómicas.
Seguimiento
Durante una mediana de seguimiento de 5 [1-20] años, 23 pacientes (64%) tuvieron al menos 1 evento arrítmico apropiado por el DAI. La mediana del tiempo hasta la aparición del primer evento fue 7 [1-29] meses. Se registraron y trataron en total 321 arritmias ventriculares de una longitud de ciclo media de 283,05 ± 35,68 ms; 7 ingresaron por tormenta arrítmica y 5 precisaron ablación de sustrato por arritmias ventriculares de repetición (3 endocárdicas y 2 epicádicas).
Todos los pacientes estaban tomando bloqueadores beta, de los que el más frecuente fue el sotalol (21 pacientes [58%]) y 11 (31%) tomaron amiodarona en algún momento.
La presencia o ausencia de variantes desmosómicas causales o su naturaleza no se asociaron con características pronósticas determinadas según la frecuencia de aparición de arritmias ventriculares o su mayor precocidad.
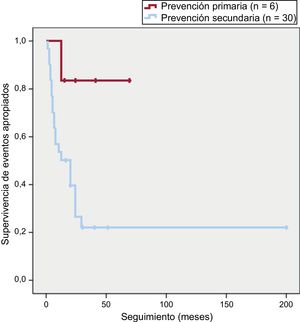
De los 23 pacientes que sufrieron eventos arrítmicos apropiados en el seguimiento, 22 (96%) eran portadores de DAI en prevención secundaria, frente a 1 en prevención primaria (p = 0,02). La supervivencia libre de eventos en función de la indicación del DAI se muestra en la figura 3.

Tuvieron eventos inapropiados durante el seguimiento 4 pacientes: 3 por fibrilación auricular paroxística y 1 por taquicardia sinusal; 3 pacientes tuvieron complicaciones relacionadas con el dispositivo: 2 infecciones y 1 rotura de electrodo.
Durante el seguimiento, 7 pacientes presentaron fibrilación auricular y 3 acudieron a urgencias por insuficiencia cardiaca. Hubo 1 trasplante cardiaco (por insuficiencia cardiaca refractaria), pero no hubo fallecidos.
DISCUSIÓNEl presente trabajo describe de manera detallada la alta incidencia de variantes desmosómicas patogénicas o probablemente patogénicas asociadas con la MCAVD de alto riesgo detectadas por medio de next-generation sequencing; además, el alto porcentaje de ellas que son mutaciones causantes de truncamiento conlleva la traducción de unas proteínas más desestructuradas.
La incidencia de mutaciones desmosómicas descritas en las series varía entre un 30 y un 60%8, como por ejemplo en la serie de den Haan et al.13, donde se detectaron variantes desmosómicas en el 52% de 82 pacientes con MCAVD, y además la presencia de estas mutaciones se asoció con mayor probabilidad de taquicardia ventricular (el 73 frente al 44%) y una edad de presentación más joven (33 frente a 41 años) en comparación con aquellos con estudio genético negativo. En la serie danesa de Christensen et al.14, se detectó un menor número de mutaciones (el 33% de 65 pacientes, 10 de ellos con diagnóstico limítrofe). La incidencia en nuestra serie es mayor, probablemente por la buena selección de pacientes, ya que todos tenían MCAVD definitiva, gran parte de ellos con arritmias ventriculares y la mitad con daño grave de la contractilidad del ventrículo derecho. A favor de ello, en un artículo reciente de Medeiros-Domingo et al.15 se detectó un mayor porcentaje de mutaciones desmosómicas en pacientes con MCAVD definitiva (n = 7) frente a MCAVD borderline o posible (n = 7), que además tenían daño de otros genes más relacionados con la miocardiopatía dilatada y se especula que serían fenocopias o cuadros solapados entre MCAVD y miocardiopatía dilatada15. Estos datos concuerdan con los del presente estudio, ya que, habiendo utilizado paneles de genes de miocardiopatías, las mutaciones detectadas eran desmosómicas y solo 4 pacientes presentaron mutaciones no desmosómicas con dudosa patogenicidad: 3 de ellas en TTN, cuyas mutaciones missense suelen tener poca transcendencia en la aparición de enfermedades y, aunque se han asociado con la MCAVD, tienen más relación con la miocardiopatía dilatada. Además, su presencia no se asoció con cambios en el pronóstico o mayor daño del ventrículo izquierdo. Estos resultados pueden llevar a pensar que en ocasiones la MCAVD no definitiva con genética negativa podría no ser una verdadera MCAVD, sino fenocopias o cuadros solapados con miocardiopatía dilatada, pero serán necesarios más estudios para confirmarlo.
Se encontraron variantes desmosómicas de nueva aparición (10) que se consideró probablemente patogénicas con base en datos de frecuencia poblacional y predictores bioinformáticos. Además, se detectó cosegregación en 6 de las 9 variantes nuevas restantes, aunque solo se incluyó en el estudio a 1 familiar (NP_004563.2: p.Gly328Glufs*24) porque cumplía los criterios de alto riesgo. El hecho de que 5 casos índice de familias diferentes presentaran la misma mutación de PKP2 (NP_004563.2: p.Glu259Glyfs*77) en la misma zona geográfica indica la existencia de un efecto fundador, lo cual puede ser una de las causas de la alta tasa de resultados genéticos positivos en esta serie. Actualmente se está realizando el estudio genético en esas 5 familias en búsqueda del probable origen común6. Por el contrario, 3 variantes detectadas se clasificaron como no asociadas con la enfermedad, por lo que profesionales expertos en genética cardiovascular deben analizar rigurosamente los resultados de next-generation sequencing para asignar su importancia y orientar el diagnóstico y el tratamiento16.
En el presente estudio las mutaciones en PKP2 se asociaron con daño exclusivamente del ventrículo derecho; por el contrario, al igual que en otros estudios, las mutaciones en DSP17 se asociaron con daño del ventrículo izquierdo (sin diferencias estadísticamente significativas).
Por otro lado, no se encontraron diferencias respecto a la presencia o ausencia de variantes patogénicas o su naturaleza en relación con un mayor número de eventos arrítmicos o su aparición más precoz. Hay que tener en cuenta que es un grupo de pacientes muy seleccionados y que la mayoría (86%) sufrió algún episodio arrítmico a lo largo de su vida, por lo que encontrar diferencias en el pronóstico parece complicado. Bhonsale et al.17 publicaron la mayor serie de pacientes con MCAVD portadores de mutaciones, y las variantes radicales no tuvieron un peor perfil arrítmico o más mortalidad o trasplantes, aunque el perfil de estos pacientes era diferente del de los nuestros, solo el 60% eran MCAVD definitivas, el 38% presentó arritmias ventriculares y el 44% eran portadores de DAI.
La importancia de la genética en esta población viene dada por la confirmación del diagnóstico (criterio diagnóstico de la Task Force4) y en el estudio familiar, con una posible detección precoz de la enfermedad, la recomendación de evitar deportes de competición, evitar pruebas diagnósticas de repetición y el estrés del seguimiento a largo plazo de pacientes con genética negativa.
El uso del DAI en este grupo de alto riesgo se justifica claramente (indicación de clase I en la guía de práctica clínica12); la mayoría de los pacientes (64%) presentaron eventos arrítmicos apropiados, pero a pesar de ello hay que destacar la ausencia de mortalidad cardiovascular y total en el seguimiento y que solo 1 paciente precisó trasplante (por insuficiencia cardiaca refractaria).
Es de interés que los portadores de DAI en prevención secundaria tuvieran una menor supervivencia libre de eventos arrítmicos que aquellos en prevención primaria. Solo 1 paciente de los 6 de prevención primaria sufrió una arritmia ventricular tratada, lo que pone de relieve la difícil selección de estos pacientes, y más aún si los factores de riesgo relacionados con un peor pronóstico arrítmico se basan en estudios retrospectivos, no aleatorizados y que en su mayoría incluyeron a pacientes que ya habían tenido un evento arrítmico mayor que motivó el implante del DAI12.
LimitacionesSe trata de un estudio retrospectivo y unicéntrico, con un pequeño número de pacientes con MCAVD de alto riesgo, que no comparó los datos con los de moderado o bajo riesgo. Además, no se incluyen estudios post mortem que también tendrían un alto riesgo arrítmico.
Dada la naturaleza retrospectiva del estudio, algún factor de confusión podría afectar los resultados obtenidos.
CONCLUSIONESLa incidencia de variantes desmosómicas patológicas o probablemente patológicas en la MCAVD definitiva de alto riesgo es muy alta y la mayoría de ellas, radicales. La presencia de dichas mutaciones o su naturaleza más desestructurada no se asociaron con características pronósticas determinadas.
Pese a tener el mismo grado de recomendación en las guías de consenso internacionales, los pacientes con MCAVD de alto riesgo en prevención primaria tienen una supervivencia libre de eventos arrítmicos significativamente mayor que aquellos en prevención secundaria.
CONFLICTO DE INTERESESNo se declara ninguno.
- –
La MCAVD es una cardiopatía definida por la sustitución progresiva de miocardio del ventrículo derecho por tejido fibroadiposo y puede dar lugar a arritmias, muerte súbita e insuficiencia cardiaca. Actualmente, un 40–60% de los pacientes muestran al menos 1 mutación genética relacionada con la enfermedad.
- –
El presente estudio detecta mediante ultrasecuenciación una alta incidencia de mutaciones en genes desmosómicos asociadas con la MCAVD definitiva por criterios diagnósticos de la Task Force y de alto riesgo por el documento de consenso de las sociedades internacionales; la mayoría de ellas causan truncamiento.
Los autores agradecen al laboratorio de genética cardiovascular Health in Code su respaldo técnico para llevar a cabo este trabajo.