
Resumir la evidencia según la cual el estrés oxidativo está en la base de diferentes procesos que conducen a la resistencia a la insulina, la inflamación y la disfunción endotelial.
Métodos y resultadosSe resalta el papel del hierro y de las citocinas inflamatorias. Se comentan las posibles ventajas terapéuticas de la depleción de hierro. Recientemente se han descubierto relaciones cuya existencia no se sospechaba entre las citocinas originadas en el tejido adiposo visceral y el metabolismo del hierro. Otros estudios han descrito relaciones divergentes entre las concentraciones de sTNRF circulantes y la función endotelial. Aunque se observó una asociación positiva entre el sTNFR1 y la vasodilatación dependiente del endotelio, se identificaron también relaciones contrarias respecto al sTNFR2, sobre todo en individuos con intolerancia a la glucosa. El conocimiento de la forma en la que se producen estas interacciones puede tener repercusiones terapéuticas. En este sentido, resulta sorprendente que la inactivación del TNFR1 en los ratones aumente la aterosclerosis y que la inactivación del NF-κβ aumente también la enfermedad vascular en los ratones. A la vista de estas observaciones paradójicas, algunos autores han planteado la posibilidad de que el sistema vascular intente limitar la captación de colesterol a través de procesos relacionados con la resistencia a la insulina.
ConclusionesEl conocimiento de los agentes que desencadenan el estrés oxidativo y la modulación de estos procesos aportarán importantes avances en la prevención y el tratamiento.
Palabras clave
La resistencia a la insulina, la intolerancia a la glucosa y la diabetes manifiesta se asocian a un aumento del riesgo de enfermedad cardiovascular1. Todos estos trastornos se acompañan de la presencia de estrés oxidativo. Se ha propuesto que éste es el mecanismo patogénico que relaciona la resistencia a la insulina con la disfunción de las células P y las alteraciones dependientes del endotelio que finalmente conducen a la diabetes manifiesta y la enfermedad cardiovascular2.
Cuando el consumo de calorías es superior al gasto de energía, el aumento de la actividad del ciclo del ácido cítrico inducido por el sustrato genera un exceso de NADH mitocondrial (mNADH) y de especies moleculares reactivas a oxígeno (ROS). Para protegerse frente a los efectos nocivos de las ROS, las células pueden reducir la formación de ROS y/o potenciar su eliminación. La evitación de la formación de ROS se logra impidiendo la acumulación de mNADH mediante la inhibición de la captación de nutrientes estimulada por la insulina y la evitación de la entrada de sustratos energéticos (piruvato, ácidos grasos) en las mitocondrias3.
El acetil-CoA, derivado de la glucosa a través del piruvato, o procedente de la oxidación beta de los ácidos grasos, se combina con el oxaloacetato para formar citrato, el cual se incorpora al ciclo del ácido cítrico y es convertido en isocitrato. La isocitrato deshidrogenasa dependiente de NAD+genera NADH. Cuando no puede eliminarse el exceso de NADH mediante fosforilación oxidativa (u otro mecanismo), el gradiente protónico mitocondrial aumenta y se transfieren electrones individuales al oxígeno, lo cual da lugar a la formación de radicales libres, y en particular de aniones superóxido.
Se ha sugerido que la interrupción de la sobreproducción de superóxido por parte de la cadena de transporte de electrones mitocondrial podría normalizar la vía que interviene en el desarrollo del estrés oxidativo4.
Recientemente se han revisado en otra publicación las repercusiones del estrés oxidativo en el desarrollo de la aterosclerosis y la diabetes tipo 22,4. Este artículo se centra en el papel de algunos desencadenantes causales del estrés oxidativo, como el hierro y las citocinas.
PAPEL DEL HIERRO EN EL ESTRÉS OXIDATIVO, LA RESISTENCIA A LA INSULINA Y LA ATEROSCLEROSISEl hierro está íntimamente relacionado con el estrés oxidativo. Participa, a través de la reacción de Fenton, en la formación de radicales libres de alta toxicidad, como los radicales superóxido e hidróxido, que son capaces de inducir una peroxidación lipídica. Para que el hierro actúe como agente prooxidante, debe estar en su forma libre. El hierro puede ser liberado de la ferritina por la acción de agentes reductores que convierten el Fe3+ en Fe2+.5 La glicación de la transferrina reduce su capacidad de unirse al hierro ferroso6 y, a través de un aumento de la cantidad de hierro libre, estimula la síntesis de ferritina. Se sabe, además, que la holotransferrina glicada facilita la producción de radicales libres de oxígeno, como el radical hidroxilo, que amplifican aún más los efectos oxidativos del hierro7.
La fracción de hierro no utilizado y altamente tóxico se almacena en forma de moléculas de ferritina con objeto de neutralizarlo. La apoferritina, que es la fracción proteica de la ferritina, está plegada espacialmente de tal manera que crea un surco central que fija las moléculas de hierro oxidadas [Fe3+]. La apoferritina es una proteína de alto peso molecular (450kDa), multimérica (24 subunidades de cadenas pesadas y ligeras), que presenta una alta y exquisita capacidad de almacenamiento de hierro (4.500 mols de hierro por mol de ferritina). La síntesis de apoferritina es inducida, a nivel tanto transcripcional como postranscripcional, por la presencia de hierro libre. El aumento de Fe2+ produce una regulación negativa de la afinidad de la proteína de fijación del elemento regulador del hierro (IRE-BP) por su lugar de fijación de IRE en la región 5' del mRNA de la ferritina, dando lugar a un aumento de la traducción de ferritina.
La cadena pesada de la molécula de apoferritina ejerce una actividad de ferroxidasa, catalizando la oxidación del Fe2+ en Fe3+, lo cual impide las reacciones redox cíclicas inducidas por el hierro que extenderían y amplificarían la lesión oxidativa. Esta actividad se produce en condiciones aerobias, y permite el almacenamiento del hierro intracelular. Cuando las concentraciones de antioxidantes son bajas, el potencial reductor y la anaerobiosis aumentan de manera progresiva, facilitando una liberación rápida del hierro de la ferritina. Además, la actividad de ferroxidasa en la cadena pesada es regulada negativamente en este contexto, con lo que se reduce la incorporación de hierro a la ferritina. El resultado global de las reacciones oxidativas es un aumento de la disponibilidad de hierro libre procedente de la molécula de ferritina, así como de otras moléculas que sufren una degradación, como el grupo hemo. Estos procesos pueden potenciar y amplificar, a su vez, el proceso de generación de radicales libres, provocando una lesión celular y tisular. El estrés oxidativo regula también negativamente la afinidad del IRE por la IRE-BP. Así pues, la ferritina puede actuar como fuente de hierro, que induce el estrés oxidativo, y también como mecanismo que protege frente a la toxicidad del hierro6.
La hiperferritinemia se da en un 6,6% de los pacientes con diabetes tipo 2 no seleccionados8. Generalmente, las concentraciones séricas de ferritina están aumentadas en los individuos con diabetes tipo 1 o tipo 2 mal controlada, y se ha demostrado que la ferritina predice los valores de HbA1c de manera independiente de la glucosa9, probablemente como consecuencia de un aumento del estrés oxidativo. Una mejora a corto plazo del control de la glucemia va seguida de reducciones variables de la concentración sérica de ferritina.
El estrés oxidativo influye también en el metabolismo de la glucosa y del hierro. Induce una resistencia a la insulina (al reducir la internalización de ésta10) y un aumento de la síntesis de ferritina.
Las citocinas pueden causar también un aumento de los receptores de transferrina en la superficie celular, favoreciendo con ello el depósito tisular de hierro11 y la resistencia a la insulina12.
En la tabla 1 aparecen las evidencias indicativas de la relación entre hierro, resistencia a la insulina y la diabetes tipo 2.
Evidencias indicativas de la relación entre hierro resistencia a la insulina diabetes tipo 2
| Hay una hiperferritinemia en un 6,6% de los pacientes con diabetes tipo 2 no seleccionados. | 8 |
| Las concentraciones séricas de ferritina suelen estar aumentadas en los pacientes con diabetes tipo 1 o tipo 2 mal controlados y se ha demostrado que la ferritina predice la HbA1c, de manera independiente de la glucosa, probablemente como consecuencia del estrés oxidativo. | 9 |
| La mejoría a corto plazo del control de la glucemia va seguida de disminuciones variables de la concentración sérica de ferritina. | 9 |
| El estrés oxidativo induce una resistencia a la insulina (al reducir la internalización de ésta) y un aumento de la síntesis de ferritina. | 10 |
| Las citocinas pueden causar también simultáneamente un aumento de los receptores de transferrina en la superficie celular, lo cual favorece el depósito de hierro y la resistencia a la insulina. | 11, 12 |
| Los radicales libres, como el O2son formados por la NADPH oxidasa y la transferencia de electrones mitocondrial. La visfatina, una nicotinamida fosforribosiltransferasa, es una enzima citosólica que interviene en la biosíntesis de NAD y presenta una asociación con los parámetros del metabolismo del hierro y la glucosa. | 18, 19 |
| La concentración máxima de mRNA de visfatina se encontró en el tejido hepático, seguido de la del tejido muscular. Estos tejidos son clásicamente sensibles a la insulina, pero se caracterizan también por desempeñar un papel central en el metabolismo del hierro. La expresión elevada de visfatina se observó también en la médula ósea, que es el origen del hierro circulante. | 13 |
| La visfatina sérica presentó una correlación negativa con la concentración sérica del receptor de transferrina soluble (sTfR). Los pacientes con una intolerancia a la glucosa y la concentración más baja de sTfR eran los que mostraban un nivel más alto de visfatina circulante. Esto sugiere que el aumento de los depósitos de hierro daría lugar a un incremento de la síntesis de visfatina. Los factores relacionados con la hiperglucemia o el estrés oxidativo pueden intervenir en parte en la asociación entre la visfatina y los parámetros del metabolismo del hierro en los individuos con una intolerancia a la glucosa. | 19 |
| Cabe prever una situación en la que la acción fisiológica de la insulina dé lugar a un aumento de la captación de diferentes nutrientes y de hierro. Cualquier factor que cause hiperinsulinemia (aumento de peso, edad, infecciones habituales repetidas, periodontitis) amplifica este proceso y determina un aumento del depósito de hierro, que a largo plazo agrava la resistencia a la insulina. | 15 |
La visfatina [también denominada factor potenciador de colonias de células pre-B13] es una nueva adipocina que es secretada predominantemente por el tejido adiposo visceral14. Como ocurre con la ferritina sérica9,15, se ha observado que la visfatina plasmática está aumentada en la diabetes tipo 2 humana16. Como se ha indicado antes, el hierro participa, a través de la reacción de Fenton, en la formación de radicales libres de alta toxicidad, como los radicales hidroxilo (OH-, a partir de Fe y H2O2), que son capaces de inducir la peroxidación lipídica17. Otros radicales libres, como el O2· son formados por la NADPH oxidasa y la transferencia de electrones mitocondriales. En este sentido, la visfatina es una nicotinamida fosforribosiltransferasa, una enzima citosólica que interviene en la biosíntesis de NAD18. La concentración máxima de mRNA de visfatina fue la observada en el tejido hepático, seguida de la del tejido muscular13. Estos tejidos son clásicamente los sensibles a la insulina, pero se caracterizan también por desempeñar un papel central en el metabolismo del hierro. Se observó también una expresión elevada de visfatina en la médula ósea, que es el origen del hierro circulante13. A la vista de esta coincidencia de la expresión de visfatina en tejidos ricos en hierro, un estudio reciente ha investigado la posible interacción de la visfatina con dicho metabolismo19.
Se observó que la visfatina sérica se asocia a diferentes parámetros del metabolismo del hierro, como la prohepcidina sérica y el receptor de transferrina soluble sérico (sTfR). Estas asociaciones diferían según el estado de obesidad y el estado de tolerancia a la glucosa. En los individuos no obesos, la concentración sérica de sTfR y la prohepcidina eran factores que contribuían de manera independiente a la variación de la visfatina tras introducir un control respecto a la edad y el IMC. Sin embargo, la sensibilidad a la insulina fue el único factor que contribuía a determinar la visfatina sérica cuando se tuvo en cuenta. En los individuos obesos, solamente el sTfR contribuía a producir la variación de la visfatina cuando se introducía un control respecto a IMC, edad, prohepcidina, sensibilidad a la insulina y estado de tolerancia a la glucosa. Estos resultados sugieren que la sensibilidad a la insulina podría ser el principal factor que influyera en las concentraciones séricas de visfatina dentro del margen completo de acción de la insulina en los individuos no obesos. Cuando se desarrolla una resistencia a la insulina (individuos obesos), el sTfR pasaría a ser uno de los factores que influyen en la concentración sérica de visfatina19.
La visfatina sérica presentaba una correlación negativa con las concentraciones séricas de sTfR. Los individuos con una alteración de la tolerancia a la glucosa y con las concentraciones más bajas de sTfR fueron los que mostraron una visfatina circulante más elevada. Esto sugiere que el aumento de las reservas de hierro daría lugar a un aumento de la síntesis de visfatina. Los factores relacionados con la hiperglucemia o el estrés oxidativo pueden intervenir en parte en la asociación existente entre la visfatina y los parámetros del metabolismo del hierro (sTfR, prohepcidina) en individuos con una alteración de la tolerancia a la glucosa19.
Así pues, la visfatina circulante parece estar relacionada con los parámetros del metabolismo del hierro, principalmente en los individuos con una alteración de la tolerancia a la glucosa19.
En resumen, puede pensarse en una situación en la que la acción fisiológica de la insulina dé lugar a un aumento de la captación de diferentes nutrientes y de hierro. Cualquier factor que cause una hiperinsulinemia (aumento de peso, envejecimiento, infecciones comunes repetidas, periodontitis) amplifica este proceso, dando lugar a un aumento del depósito de hierro, que a largo plazo agrava la resistencia a la insulina.
DEPLECIÓN DE HIERRO Y ATEROSCLEROSISEl efecto general del hierro catalítico consiste en convertir los radicales libres poco reactivos, como H2O2, en otros altamente reactivos (OH- y O2-). Los radicales libres y otros productos derivados de la oxidación son factores conocidos que deterioran los mecanismos de vasodilatación20 y causan una depleción endotelial de antioxidantes endógenos, como el ácido ascórbico21. La quelación del hierro bloquea la oxidación de las lipoproteínas de baja densidad y el hierro liberado del grupo hemo y la ferritina favorece la oxidación de estas lipoproteínas22. Así pues, teóricamente, es de prever que el aumento de disponibilidad de hierro contribuya a producir la enfermedad macrovascular, puesto que el hierro ejerce efectos adversos sobre el endotelio23 y acelera el desarrollo de la aterosclerosis24. De hecho, la expresión del gen de la ferritina aumenta en el transcurso de la formación de la placa aterosclerótica25.
En los individuos con hemocromatosis, las arterias de tamaño medio se caracterizan por una hipertrofia excéntrica y una disminución de la distensibilidad, que son en parte reversibles tras una depleción de hierro26. Estas observaciones parecen estar relacionadas con la fibrogénesis inducida por el hierro que determina un aumento del contenido total de colágeno en las arterias de estos pacientes. Hay también algunos datos que indican un crecimiento del tejido de la pared arterial dependiente del hierro: la quelación del hierro mediante deferoxamina inhibe la proliferación de las células de músculo liso vascular27.
El uso a largo plazo del quelante de hierro modificado deferoxamina conjugada con hidroxietilalmidón previno la disfunción endotelial asociada a la diabetes mellitus experimental28. En pacientes con diabetes tipo 2, las respuestas arteriales coronarias a la prueba de estrés por frío mejoraron de manera sustancial tras la administración de deferoxamina29. De forma análoga, se demostró que la quelación del hierro aportaba una mejora en la disfunción endotelial de los pacientes con enfermedad coronaria30.
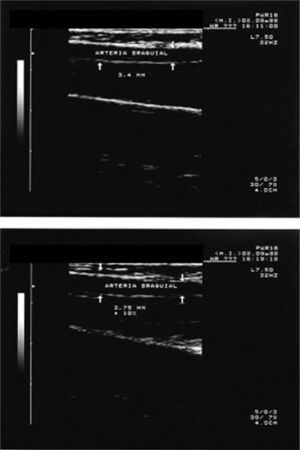
Se observó asimismo una mejora de la vasodilatación inducida por nitroglicerina tras la extracción de sangre en pacientes con diabetes tipo 2. La mejora de la reactividad vascular fue paralela al descenso de la saturación de transferina sérica, la hemoglobina total (marcadores del hierro circulante) y la hemoglobina glicada de la sangre31. Estas observaciones sugieren que la disfunción vascular diabética parece ser parcialmente reversible y que el compartimento circulante actúa como un reservorio de metales de transición que afecta directamente a la función vascular32. El aumento de la hemoglobina, una proteína rica en hierro, es nocivo para la función endotelial, puesto que se sabe que los vasos sanguíneos normales expuestos a la hemoglobina total y a la hemoglobina glicada experimentan un deterioro de la relajación vascular33 (fig. 1).
 y después (panel inferior) de una vasodilatación inducida por isquemia.")
Sin embargo, la relación entre el hierro y la aterosclerosis es motivo de controversia34. Así, algunos datos experimentales obtenidos en animales respecto al efecto de la manipulación de las reservas de hierro sobre la aterosclerosis, y los datos obtenidos en el ser humano que indican una mejora de la estructura y la función vasculares tras una depleción de hierro26,29–31, concuerdan con la teoría de que el hierro contribuye al desarrollo de la enfermedad vascular. No obstante, los datos epidemiológicos actuales que relacionan los depósitos de hierro con la aterosclerosis o con la enfermedad coronaria34 no respaldan plenamente esta hipótesis.
En resumen, el impacto de los metales de transición en general y del hierro en particular, sobre la fisiología humana tan sólo ha empezado a aclararse en las últimas décadas. Dado que el hierro es un prooxidante de primera línea, contribuye a regular las manifestaciones clínicas de numerosas enfermedades sistémicas, incluida la diabetes mellitus y la aterosclerosis. La regulación que ejerce el hierro en el estrés oxidativo celular puede explicar, al menos en parte, su estrecha asociación con las anomalías de la sensibilidad a la insulina.
citocinas, estrés oxidativo y enfermedad cardiovascularDiferentes citocinas inducen alteraciones mitocondriales y un aumento de la producción de radicales libres. Las citocinas desempeñan también un importante papel en la lesión endotelial inducida por la inflamación. El endotelio vascular interviene en la respuesta inflamatoria a la aterosclerosis35–38, y los cambios de la función endotelial podrían estar en la base de la asociación entre enfermedad cardiovascular e inflamación.
En los últimos años, se ha obtenido una evidencia creciente que indica que la inflamación crónica está relacionada simultáneamente con la disfunción endotelial, la aterosclerosis y la resistencia a la insulina39,40. La disfunción endotelial es una de las primeras anomalías que pueden detectarse en los individuos con riesgo de sufrir episodios cardiovasculares y está relacionada con la resistencia a la insulina y la diabetes tipo 236,37. Las concentraciones plasmáticas de citocinas proinflamatorias como la interleucina (IL)-18, la IL-6 y el factor de necrosis tumoral alfa (TNF-α) y de otros varios marcadores inflamatorios están aumentadas en los pacientes con cardiopatía isquémica39,41–43. Los varones que han estado expuestos a un aumento de las proteínas plasmáticas sensibles a la inflamación presentan una letalidad superior en futuros episodios coronarios, después de introducir un ajuste para los factores de riesgo tradicionales41,44. Las citocinas circulantes están también elevadas en la diabetes tipo 2, la obesidad y el síndrome de resistencia a la insulina y desempeñan un papel central en la patogenia de estos trastornos39.
InterleucinasConcretamente, se ha afirmado que la liberación de IL-6, principalmente procedente de los adipocitos abdominales, desempeña un papel clave en la relación entre el estrés oxidativo y la disfunción endotelial3. La IL-6 es un mediador de la respuesta inflamatoria y está ligada a la dislipidemia, la diabetes tipo 2 y el riesgo de infarto de miocardio39,45–47. La IL-6 es secretada por diversos tipos celulares diferentes, incluidas las células linfoides y endoteliales, los fibroblastos, el músculo esquelético y el tejido adiposo. Las concentraciones de IL-6 circulante están correlacionadas también con la obesidad y la resistencia a la insulina, y pueden predecir la aparición de una diabetes tipo 247–50.
Resulta difícil determinar si las citocinas inducen la lesión endotelial de forma directa o, al menos en parte, a través de la inflamación asociada a la resistencia a la insulina. Recientemente, se ha descrito una asociación independiente entre la resistencia a la insulina y la disfunción vascular en 81 pacientes con diabetes tipo 237. La resistencia a la insulina se acompañó de una disminución de la vasodilatación dependiente del endotelio y un aumento de la inflamación de bajo grado. En la diabetes tipo 2, se ha sugerido también la presencia de una sensibilidad característica del endotelio a la resistencia a la insulina37.
En otro estudio, se observó una correlación negativa entre las concentraciones de IL-6 y la vasodilatación dependiente del endotelio, que continuaba siendo significativa tras introducir un ajuste respecto a la sensibilidad a la insulina y otros factores de riesgo, en varones aparentemente sanos51. En dicho estudio los autores analizaron a varones sanos con objeto de reducir al mínimo el efecto de confusión producido por otros factores de riesgo o tratamientos que pudieran interferir en la interpretación de los resultados51. La asociación entre las concentraciones séricas de IL-6 y la vasodilatación dependiente del endotelio fue especialmente significativa en los individuos con una glucosa en ayunas estrictamente normal51. Hay otros factores y citocinas en el contexto de la hiperglucemia y la resistencia a la insulina que pueden atenuar la relación entre la IL-6 y la disfunción endotelial cuando aumenta la glucosa en ayunas.
Se ha demostrado que las células del endotelio y del músculo liso producen IL-652. La inflamación puede producir una disfunción endotelial a través de diferentes mecanismos. La inflamación es capaz de deteriorar la vasodilatación mediada por el flujo tanto a través de un aumento de la vasoconstricción como por una reducción de los vasodilatadores de origen endotelial53. Las citocinas pueden inducir también una vasoconstricción a través de diferentes vías, como la inducción de la síntesis de endotelina-1, la reducción de la expresión de óxido nítrico (NO) sintasa endotelial o la disminución de la biodisponibilidad del NO40.
En el estudio de la segunda generación de Framingham, se demostró una correlación inversa entre las concentraciones de IL-6 y la función endotelial, y esta relación se atenuaba tras un ajuste respecto a los factores de riesgo tradicionales54. La relación entre la IL-6 y la vasodilatación mediada por el flujo se ha descrito también en individuos con síndrome coronario agudo e hipercolesterolemia55,56. En esos estudios, a pesar de la estrecha relación existente entre el síndrome metabólico y el riesgo cardiovascular, no se evaluó el efecto de la resistencia a la insulina sobre la función endotelial.
Se ha demostrado que la rosiglitazona mejora la reactividad vascular de la misma forma que reduce las concentraciones de IL-6, lo cual sugiere un papel de esta citocina en el efecto de la resistencia a la insulina sobre la función vascular57.
Otra hipótesis explicativa sería que una inflamación de bajo grado (aumento de interleucina 6) pudiera constituir la agresión inicial, la cual ejercería un papel clave en la agrupación de inflamación/resistencia a la insulina, al inducir predominantemente una disfunción endotelial. En una segunda agresión posterior, el efecto de la resistencia a la insulina sobre el endotelio pasaría a ser más importante de por sí, y desempeñaría el papel central en las anomalías metabólicas asociadas a la disfunción vascular51. Serán necesarios estudios prospectivos que analicen simultáneamente la inflamación, la resistencia a la insulina y la función endotelial para comprender mejor el mecanismo que subyace en estos procesos.
Debe reconocerse también que la respuesta inflamatoria crónica en el contexto de la resistencia a la insulina y la disfunción endotelial se produce no sólo con la interleucina 6 sino también probablemente con multitud de otros factores. En futuros estudios, será interesante determinar si la interleucina 6 es más importante que otras citocinas y factores circulantes.
PAPEL DEL FACTOR DE NECROSIS TUMORAL ALFA (TNF-α)El TNF-α es una citocina proinflamatoria que interviene también en la patogenia de la resistencia a la insulina y en la disfunción del endotelio ligada a este fenómeno39,58. En diferentes estudios se han descrito efectos contradictorios del TNF-α sobre la función endotelial59–61. La infusión intrabraquial aguda de TNF-α deteriora la vasodilatación dependiente del endotelio, pero el TNF-α potencia también el mecanismo de protección58–61.
El TNF-α lleva a cabo sus múltiples actividades biológicas tras la unión a dos receptores de membrana diferentes, el TNFR1 y el TNFR2, activando diferentes cascadas de señalización, con lo que facilita respuestas celulares distintas62. Tras la unión a estos receptores, una fragmentación proteolítica de las partes extracelulares da origen a las formas solubles, denominadas sTNFR1 y sTNFR263. Se cree que las concentraciones de sTNFR1 y sTNFR2 reflejan los efectos previos del TNF-α63.
El desprendimiento de TNFR1 da lugar a un aumento de sTNFR1, que antagoniza el TNF-α64. El aumento de la expresión de sTNFR1 redujo la bioactividad de TNF-α y protegió el miocardio frente al infarto tras la isquemia y reperfusión65,66. El sTNFR1 podría tener otras funciones protectoras a través de la estimulación del crecimiento de las células endoteliales67. Por otra parte, las concentraciones de sTNFR2 se han relacionado con la enfermedad coronaria68, la resistencia a la insulina69 y la hipertensión70.
Los estados de resistencia a la insulina se asocian también a un deterioro del desprendimiento de sTNFR1 y un aumento del desprendimiento de sTNFR270,71. La regulación positiva mantenida del TNFR2 humano en ratones transgénicos conduce a una acumulación crónica de receptores de la superficie celular y plasmáticos72, que les proporciona una capacidad de hiperrespuesta al TNF-α circulante.
Estos mecanismos antiateroscleróticos inducidos por los sTNFR1, y los mecanismos proateroscleróticos asociados al sTNFR2 concuerdan con algunas observaciones recientes73. Se evaluó la relación entre los receptores de TNF-α soluble, la sensibilidad a la insulina y la reactividad vascular en individuos con una tolerancia a la glucosa normal o alterada. Se observó una correlación positiva entre las concentraciones de sTNFR1 y la vasodilatación dependiente del endotelio (r=0,291, p=0,02) en los individuos con una tolerancia a la glucosa normal. En un análisis de regresión múltiple, el sTNFR1 en suero contribuía de manera independiente a la varianza de la vasodilatación dependiente del endotelio (VDDE) en estos individuos, tras introducir un ajuste respecto a edad, IMC, tabaquismo, presión arterial sistólica y diastólica y sensibilidad a la insulina (B=0,414, p=0,002)73. El sTNFR2 circulante presentaba una asociación negativa con la sensibilidad a la insulina (r=−0,20, p=0,04) y se observó una tendencia respecto a la VDDE (r=−0,190, p=0,058). En los individuos con intolerancia a la glucosa, las concentraciones séricas de sTNFR2 presentaban una correlación negativa con la VDDE (r=−0,366, p=0,047). Sin embargo, la relación no era significativa tras introducir un ajuste respecto a variables de confusión73. No se observó asociación alguna entre la vasodilatación independiente del endotelio y las concentraciones de sTNFR1 o sTNFR2 circulantes73.
Este estudio ha mostrado, pues, relaciones divergentes entre las concentraciones de sTNFR circulante y la función endotelial. Aunque el sTNFR1 tenía una asociación positiva con la VDDE, se observó la relación contraria con el sTNFR2, principalmente en los individuos con intolerancia a la glucosa73. El conocimiento de la forma en la que se producen estas interacciones puede tener repercusiones terapéuticas. A este respecto, resulta sorprendente que la inactivación de un TNFR1 en los ratones aumente la aterosclerosis74. La inactivación de NF-κβ también aumenta la enfermedad vascular en el ratón75. A la vista de estas observaciones paradójicas, se ha planteado la posibilidad de que el sistema vascular pueda intentar limitar la captación de colesterol a través de procesos relacionados con la resistencia a la insulina76.
CONCLUSIONESEn resumen, se están identificando cada vez mejor los agentes desencadenantes que conducen al estrés oxidativo (hierro y citocinas inflamatorias) y la modulación de estos procesos. El conocimiento de estas interacciones aportará, indudablemente, un rendimiento preventivo y terapéutico.