La miocardiopatía arritmogénica (MCA) es un trastorno hereditario que se caracteriza por el reemplazo progresivo del miocardio del ventrículo derecho (VD) y el ventrículo izquierdo (VI) por tejido fibroadiposo, lo que implica un retraso en la conducción del impulso eléctrico y el desarrollo de arritmias ventriculares malignas. Se trata de una de las principales etiologías de la muerte súbita cardiaca (MSC) de individuos jóvenes y aparentemente sanos. A nivel celular, en los últimos años, se han realizado avances sustanciales en cuanto a sus mecanismos genéticos y fisiopatológicos, específicamente en dianas genéticas no desmosomales que afectan al VI. En la época de la secuenciación de nueva generación (NGS), se ha implicado a más de 15 genes en la etiopatogenia de la MCA, con una vía patológica final común de muerte celular y fibrosis1. De hecho, entre los criterios diagnósticos actuales del grupo de trabajo de 2010 se incluye la presencia de una mutación patógena clasificada como asociada o probablemente asociada con la MCA. Aunque clásicamente la MCA se ha relacionado con mutaciones que afectan directamente a las proteínas desmosomales, la genética ha contribuido a identificar un espectro de la enfermedad mucho más amplio y ha extendido su espectro genético más allá de los genes desmosomales. Ejemplos de genes no desmosomales asociados con signos clínicos y anatomopatológicos clave de la MCA del VI son, fundamentalmente, genes que codifican proteínas del citoesqueleto (DES), la membrana nuclear (LMNA o TMEM43), el retículo sarcoplásmico (PLN) o el disco Z (FLNC)1.
Esto último hace que se plantee la pregunta de cómo una molécula de la membrana nuclear o del citoesqueleto genera un fenotipo de MCA con afección del VD o principalmente del VI o biventricular con un comportamiento eléctrico anómalo. Los miocardiocitos crean conexiones estructurales y eléctricas por medio de desmosomas, uniones de adhesión y uniones intercelulares comunicantes, que conforman uniones de tipo mixto, conocidas como área composita, ubicadas en los discos intercalares. Una función importante de los desmosomas es conectar mecánicamente las células adyacentes uniendo sus filamentos intermedios para crear una red citoesquelética unificada. Mediante estas interacciones, la desmina coordina el movimiento de los discos Z vecinos con las membranas nuclear y plasmática3. Por consiguiente, esta red mecánica está estrechamente relacionada con la filamina C en los discos Z y el complejo LINC (conector del nucleoesqueleto y el citoesqueleto) compuesto por laminas y proteínas LUMA (TMEM43), así como por canales iónicos de la membrana plasmática, como el canal de sodio NaV1.5, entre otros. Por lo tanto, fallos en esta red pueden alterar la integridad estructural y la mecanotransducción de los miocardiocitos, y serían el posible mecanismo subyacente a las mutaciones desmosomales en la MCA. Aún hay importantes lagunas en el conocimiento sobre la patogénesis molecular de este cuadro clínico, pero es posible que el fenotipo de la MCA sea la expresión común de una cascada de errores en este complejo macromolecular desde el sarcolema y el área composita hasta el núcleo.
El diagnóstico de MCA representa todo un reto y se basa en la combinación de datos eléctricos, estructurales, genéticos y anatomopatológicos, así como en los antecedentes familiares. Los criterios diagnósticos del grupo de trabajo, publicados por primera vez en 1994 y actualizados después en 20102, combinan todos estos datos y requieren 2 criterios mayores o 1 criterio mayor y 2 criterios menores o 4 criterios menores para el diagnóstico definitivo de la MCA. El diagnóstico definitivo de la MCA es especialmente complejo debido a las variables penetrancia y expresión de la enfermedad, incluso entre los miembros de una misma familia con una mutación común, con una incidencia relativamente frecuente de portadores silentes. Para detectar cambios sutiles en el miocardio que indiquen MCA, las técnicas de imagen cardiaca han evolucionado mucho en la última década. Por ello, la imagen cardiaca avanzada debería ser una de las piedras angulares en el diagnóstico de la MCA; la resonancia magnética cardiaca (RMC) es la técnica más sensible no solo para la definición de la estructura cardiaca, sino también para su caracterización tisular.
La RMC es cada vez más relevante en el campo de las miocardiopatías isquémica y no isquémica, ya sea por una mejor caracterización de la enfermedad al proporcionar datos clave en el diagnóstico diferencial o mejorando su estratificación pronóstica4. De especial interés es el valor diagnóstico de la RMC en otras miocardiopatías no isquémicas, y se han propuesto patrones específicos de realce tardío del gadolinio (RTG) en diversas situaciones clínicas5. Además, la extensión del RTG parece correlacionarse bien con la presencia de eventos arrítmicos adversos, como la MSC y las miocardiopatías dilatada e hipertrófica. Sin embargo, aún no se dispone de datos sólidos sobre el papel de la RMC en la MCA de predominio izquierdo.
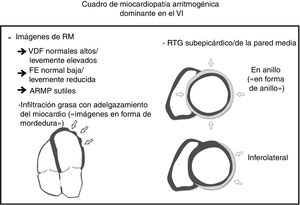
En un artículo publicado recientemente en Revista Española de Cardiología, Feliu et al.6 presentan un impecable estudio que intenta profundizar en el conocimiento sobre el valor diagnóstico y pronóstico de la RMC en la MCA izquierda. Los autores deben congratularse por un trabajo brillante que ayuda a resaltar el papel de la RMC en la MCA. Con un tamaño de muestra suficiente, pudieron establecer una correlación entre el RTG del VI extenso con un peor pronóstico, causado por eventos arrítmicos graves o insuficiencia cardiaca. Además, muestran un hallazgo característico común en las RMC, al que denominan «signo de la mordedura de rata», en un importante porcentaje de los casos, como resultado de la infiltración grasa (IG) del VI, que es muy indicativo de MCA. Por último, sus resultados confirman los patrones previamente descritos de RTG inferolateral-subepicárdico y circunferencial como una pista diagnóstica típica de la MCA.
Desde una perspectiva clínica, es probable que esta muestra esté sobrerrepresentada con casos en fases clínicas avanzadas. De hecho, el 80% de los casos incluidos son probandos, la mayoría de ellos ya habían tenido antes un episodio de arritmia ventricular sostenida o eran portadores de un desfibrilador automático en prevención secundaria de MSC. Esto puede explicar la tasa relativamente elevada de eventos adversos observados. Además, la naturaleza mixta del diseño del estudio, considerando el evento cardiaco arrítmico inicial para el análisis estadístico de seguimiento, aporta un sesgo en la interpretación del resultado clínico real. En muestras prospectivas mayores, como la de Mazzanti7, la tasa de eventos cardiacos adversos a los 5 años fue considerablemente menor. Esta naturaleza ambispectiva de la muestra de pacientes en el estudio de Feliu et al. puede limitar las conclusiones sobre la elevada prevalencia de los hallazgos en la RMC, como el «signo de la mordedura de rata» o el RTG inferolateral extenso, que podría no encontrarse en familiares y portadores de mutaciones en una fase más preclínica. Esto aún constituye el principal reto diagnóstico en la MCA para alcanzar un diagnóstico precoz.
Coincidimos con Feliu et al.6 en que las formas predominantes del VI y biventricular de la MCA están infrarrepresentadas en los criterios de RMC del grupo de trabajo de 20102, y una futura actualización debería incluir ambos fenotipos. Está claramente reconocida la frecuente afección del VI en la MCA. En consonancia con datos previos, nuestro grupo también ha hallado anomalías regionales de la contractilidad parietal o RTG en hasta el 79,5% de los casos8, y contribuciones recientes han demostrado que hasta el 70% de las MSC en las autopsias de MCA han mostrado afección biventricular en el análisis histopatológico9. En los últimos años, ha crecido el interés por este fenotipo del VI. A falta de criterios actualizados, los estudios recientes en RMC han mostrado algunas características que podrían ayudar a comprender las formas biventricular y predominante izquierda. Inicialmente, Sen-Chowdhry et al.10 propusieron como rasgos diagnósticos la presencia de aneurismas del VI, una ligera dilatación del VI, la alteración sistólica y el RTG con distribución subepicárdica/mesocárdica. Sin duda, en la literatura, la afección del VI por RTG se describe en las capas subepicárdica/mesocárdica de la pared, hallazgo que se ha correlacionado histopatológicamente al mostrar infiltración fibroadiposa con un gradiente característico desde el estrato externo hacia el interno («fenómeno del frente de onda»), que afecta a las capas subepicárdicas con cierta extensión al mesocardio11.
Los criterios del grupo de trabajo de 2010 reconocen que el reemplazo por tejido fibroadiposo2, y no solo la IG, debería ser el verdadero sello distintivo de la enfermedad (figura 1). Las secuencias de la RMC con supresión de la grasa tienen varias limitaciones, como la alta variabilidad interobservador, la dificultad en su interpretación y su baja especificidad. Es importante tener en cuenta que la IG puede observarse en otros supuestos clínicos. Se puede encontrar IG en el VD en hasta el 50% de los corazones normales de ancianos, con miocitos que parecen desplazarse en lugar de reemplazarse, sin evidencia de fibrosis, degeneración de miocitos o inflamación12. Además, un aumento de tejido graso en el subepicardio es un hallazgo normal en personas obesas (adipositas cordis) y no debe interpretarse erróneamente como MCA. Por último, se puede observar IG en otras entidades, como tras un infarto de miocardio, en lipomas cardiacos, en esclerosis tuberosa y en la miocardiopatía dilatada13, En la MCA, la IG se ha descrito anteriormente tanto en el VD como en el VI como proyecciones «digitiformes» que alteran el contorno normal de la pared; sus ubicaciones preferentes son las paredes epicárdicas libres del VD y el VI13, lo que está en concordancia con los hallazgos de Feliu et al.6. El novedoso «signo de la mordedura de rata» descrito por estos autores es un hallazgo sugerente y valioso que hace sospechar firmemente en una MCA preexistente. Sin embargo, la ausencia de IG en algunas formas de la enfermedad, como el síndrome de Carvajal (que frecuentemente se asocia con RTG), reduce su sensibilidad como marcador diagnóstico de la MCA14.

El amplio espectro de la enfermedad se puede expresar de diferentes formas y el estado genético subyacente podría desempeñar un papel importante. De hecho, hemos observado que los pacientes con mutación en genes desmosomales se manifiestan principalmente con la forma típica de miocardiopatía arritmogénica del VD con o sin fenotipo biventricular, mientras que mutaciones en genes no desmosomales se expresan como formas de VI y biventricular8. Estudios recientes han demostrado que no solo la presencia de RTG, sino también su extensión y su localización inferolateral/subepicárdica, son importantes factores de riesgo pronóstico5. Creemos que la distribución del RTG puede depender del trastorno genético, ya que hemos observado que los portadores de la mutación de FLNC han mostrado preferencia por la pared inferolateral, mientras que la mutación en desmina ha presentado un patrón anular subepicárdico extenso característico (patrón en anillo)8. De manera semejante a la IG, estos patrones de RTG, aunque no son patognomónicos, son muy indicativos de MCA. Por lo tanto, el RTG, como signo distintivo del reemplazo fibrótico, debe desempeñar un papel esencial en los próximos criterios de la MCA, aunque claves como la IG con adelgazamiento miocárdico, volúmenes del VI ligeramente aumentados, fracción de eyección ligeramente reducida o anomalías sutiles de la contractilidad regional parietal pueden contribuir al diagnóstico definitivo (figura 1).
Por último, Feliu et al. han identificado algunos predictores clínicos en la estratificación del riesgo que empeoran el pronóstico, como el sexo masculino y la práctica deportiva6. El sexo se ha propuesto como un marcador de riesgo arrítmico en varios subtipos genéticos específicos de MCA, como LMNA,FLNC,TMEM43 y DES15. En estudios previos, en una muestra más heterogénea de pacientes con MCA del VD también se ha encontrado una correlación positiva entre el sexo masculino y un peor pronóstico, y los datos de Feliu et al. sirven para confirmar este hallazgo. En cuanto al deporte, aunque este artículo no perseguía este objetivo específico, los resultados continúan siendo controvertidos. En estudios anteriores se ha demostrado claramente que el riesgo arrítmico estaba directamente relacionado con la práctica previa de deportes de competición. Sin embargo, la mayoría de los estudios en este ámbito no son del todo exactos en lo que respecta al registro de la actividad física y, hasta la fecha, los consejos específicos sobre el deporte no deberían recomendar categóricamente evitar cualquier tipo de práctica deportiva.
En resumen, estamos de acuerdo con los autores en que es hora de actualizar los criterios diagnósticos de la MCA. Se dispone de datos recientes sobre caracterización tisular del miocardio y patrones de RTG mediante imagen cardiaca, así como sobre un subconjunto de genotipos de alto riesgo con apetencia conocida por el VI, específicamente por el subepicardio inferolateral. Los cardiólogos de todo el mundo ya esperan una revisión de los criterios diagnósticos que integren todos estos avances y den paso a estrategias de tratamiento adecuadas.
CONFLICTO DE INTERESESLos autores no declaran ningún conflicto de intereses.